





X-linked hypophosphatemia (XLH) is the most prevalent form of hereditary rickets. It is caused by loss-of-function mutations in the phosphate-regulating Endopeptidase Homolog X-linked (PHEX) gene, which results in elevated circulating levels of fibroblast growth factor 23 (FGF23), which, in turn, gives rise to a high waste of phosphate in urine, low serum phosphate concentration, and reduced production of 1,25 dihydroxy vitamin D [1,25(OH)2 vitamin D], the active form of vitamin D in our body.
XLH is transmitted following a dominant inheritance linked to chromosome X. Thus, mothers with XLH transmit the disease to 50% of their offspring, and fathers with XLH transmit the disease to all their daughters and none of their sons. However, it is of note that in the majority of cases, the PHEX gene mutations appear de novo, that is, the patient has the disease, but the parents are healthy. It is not known why new mutations arise.
Though the severity of clinical manifestations is highly variable, children with XLH characteristically present with vitamin D-resistant rickets (Figure 1) during infancy and develop shortening of lower extremities, bone deformities (Figure 2), and disharmonic growth failure.
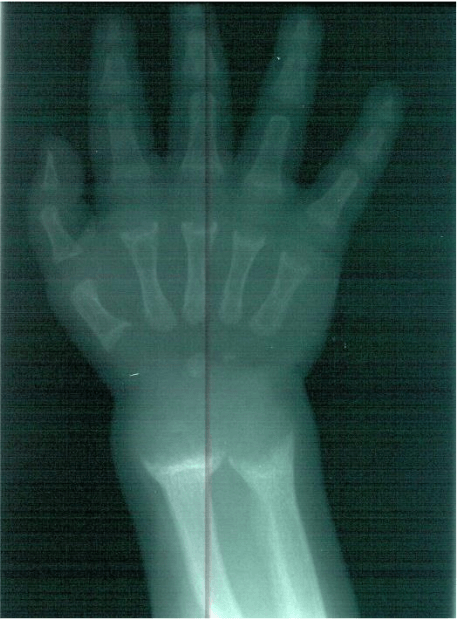
Figure 1: Radiological widening of the wrist (metaphyseal cupping), due to the protrusion of bulky mass of cartilaginous cells in the zone of hypertrophy into the poorly mineralized metaphysis. Image courtesy of the authors.
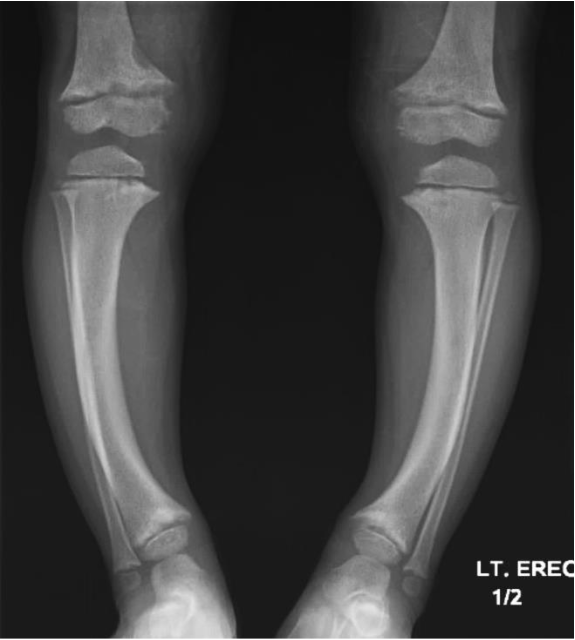
Figure 2: X-ray of legs showing bowing of tibiae (the weight-bearing bones). Image courtesy of the authors.
Adults have reduced final height, leg bowing (Figure 3), and bones deficiently mineralized (osteomalacia) that may cause bone pain and muscular weakness. Dental abnormalities are frequently found in children and adults, and patients with XLH may also develop deafness and vertigo in the long term if the bones of the inner ear are involved.

Figure 3. Bowing of both legs – a classical sign of rickets. Image courtesy of the authors.
Maintained oral phosphate supplementation and administration of vitamin D derivatives ameliorate the manifestations of XLH, and this has been the basis of conventional, classic therapy of XLH. However, this combined treatment does not decrease the elevated FGF23 levels or antagonize other potential undesirable actions of FGF23. This is important because high FGF23 has been shown to induce left ventricular hypertrophy (enlargement of heart muscle cells),1 and it has been related with cardiac toxicity and the increased risk of death by cardiovascular complications in patients with chronic kidney disease.2 Thus, it could be hypothesized that patients with XLH, who keep elevated serum concentrations of FGF23 despite treatment, might develop cardiovascular damage, even in childhood.
To address this issue, we studied 24 children from 1 to 17 years of age (17 females) diagnosed with XLH and harboring PHEX mutations.3 Patients had been enrolled in the RenalTube database (www.renaltube.com) (Figure 4) and were receiving treatment with phosphate and vitamin D derivatives. In addition to biochemical parameters including determination of serum FGF23 values, we analyzed 24 hours of ambulatory blood pressure monitoring (ABPM), carotid vessel ultrasonography to assess intima-media thickness (broadness of vessel wall), and doppler echocardiography. By doing this, we wanted to find out whether these patients had signs of cardiovascular toxicity demonstrated by arterial hypertension, arteriosclerosis, and/or left ventricular hypertrophy.

Figure 4: Renal tube database – for the study of primary tubulopathies. Figure courtesy of the authors.
All but one patient had serum FGF23 concentrations above the normal reference values. Abnormally high carotid intima-media thickness and arterial hypertension were found in one case, but this finding was attributed to the marked obesity of this patient rather than XLH. Arterial hypertension was disclosed in two other children. The group’s mean left ventricular mass index (LVMI) measured by cardiac ultrasonography was 44.93± 19.18 g/m2.7, and four patients, in addition to the obese one, had values greater than 51 g/m2.7, a value considered to be indicative of left ventricular hypertrophy. There was no correlation between these echocardiographic parameters and serum FGF23 concentrations.
In summary, our results showed that XLH pediatric patients receiving conventional treatment have echocardiographic measurements of ventricular mass within normal reference values, but above the mean, and 18% have LVMI suggestive of left ventricular hypertrophy without correlation with serum FGF23 concentrations. This might indicate an increased risk of cardiovascular involvement in XLH since an early age.
These findings have important clinical implications and support the need for monitoring cardiovascular disease in the diagnosis and follow-up of patients with XLH. Moreover, the possibility of increased cardiovascular toxicity in XLH raises the issue of whether treatment with burosumab, a new monoclonal antibody against FGF23 shown to cause beneficial metabolic and clinical effects in XLH 4, might also be useful in preventing or reversing left ventricular hypertrophy in these patients.
Further research studies are necessary to clarify the risk of cardiovascular involvement in XLH and the actual pathogenic role of FGF23 as well as the potential positive consequences of burosumab treatment on this complication of XLH.
Related Posts
 Important Strategies To Help Healthcare Providers Support Patients With Diabetes
Important Strategies To Help Healthcare Providers Support Patients With Diabetes Studying The Link Between Increased BMI And Late-Onset Preeclampsia In Pregnant Women
Studying The Link Between Increased BMI And Late-Onset Preeclampsia In Pregnant Women A Bacterial Cell Imaging Method Using CRISPR And Microfluidics
A Bacterial Cell Imaging Method Using CRISPR And Microfluidics B. infantis Reduces Key Markers Of Intestinal Inflammation In Infants
B. infantis Reduces Key Markers Of Intestinal Inflammation In Infants Epigenetic Changes In Multiple Sclerosis – Studied In Twins
Epigenetic Changes In Multiple Sclerosis – Studied In Twins Did You Know Math Can Help Us Learn How Diseases Work?
Did You Know Math Can Help Us Learn How Diseases Work?