





As a family of epigenetic enzymes, histone deacetylases (HDACs) can catalyze the removal of an acetyl group from N-acetyl lysine residues of histones and other proteins. The human HDACs can be grouped into 4 classes based on their homology to yeast prototypes: Class I (HDAC1, 2, 3 and 8), Class II (HDAC 4,5, 6, 7, 9 and 10) and Class IV (HDAC11) are zinc-dependent, while Class III (SIRT1-7) is NAD+-dependent.
HDAC8 is a unique zinc-dependent class I HDAC, identified as a 42 kDa protein composed of 377 amino acids and located in the nucleus and cytoplasm, where it plays numerous physiological and pathological roles. More importantly, there is increasing evidence showing that HDAC8 aberration contributes essentially to the onset and progression of many diseases, especially T-cell lymphoma, childhood neuroblastoma and Cornelia de Lange Syndrome (CdLS). Therefore, development of HDAC8 selective inhibitors is of great therapeutic value.
In the present research, the knowledge-based rational design of HDAC inhibitor led to the discovery of three meta-sulfamoyl N-hydroxybenzamide-based HDAC8 selective inhibitors 12a-12c (Figure 1), which exhibited potent HDAC8 inhibition with two-digit nanomolar IC50 values, and remarkable selectivity over HDAC2 and HDAC6. Additionally, western blot analysis also supported such selectivity on the intracellular level.
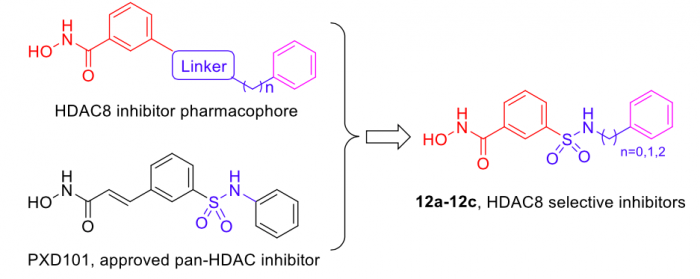
Figure 1. Design of meta-sulfamoyl N-hydroxybenzamides-based HDAC8 inhibitors. Credit: Yingjie Zhang
It’s worth noting that compounds 12a-12c possessed selective cytotoxicity to T-cell leukemia cell lines Jurkat, Molt-4 and neuroblastoma cell line SK-N-BE-(2), which was also observed in the well-known HDAC8 selective inhibitor PCI-34051. The molecular docking study showed compounds 12a (Fig 2A), 12b (Fig 2B), 12c (Fig 2C), and PCI-34051(Fig 2D) shared a similar binding mode in the active site of HDAC8.
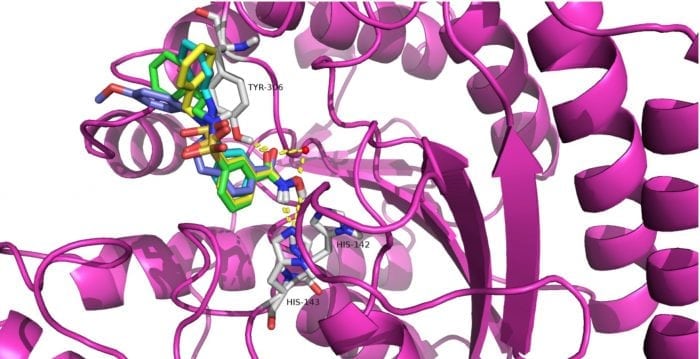
Figure 2. Proposed binding model of compounds 12a (cyans), 12b (yellow), 12c (green) and PCI-34051 (blue) with HDAC8 (PDB code: 2V5X). The zinc ion is shown as a red sphere. Hydrogen bonds and coordination of the zinc ion are shown as yellow dashed lines. Credit: Yingjie Zhang
In the past decades, several hundreds of clinical trials of more than 20 HDAC inhibitors, mainly as antitumor agents, have been initiated. However, most HDACIs in the clinic are pan-HDAC inhibitors or class selective inhibitors, which inhibit broad-spectrum HDACs and lead to nonnegligible toxicities. Therefore, development of HDAC inhibitors selective for a single isoform with the expectation of better tolerance and fewer side effects has become a hot topic. Though many HDAC8 selective inhibitors have been developed and exhibited satisfying toxic profiles, their in vitro and in vivo antitumor potency were not compelling enough for further clinical evaluation.
Two key points for the development of HDAC8-selective inhibitors are to find the accurate disease model and to get antitumor synergy in combination with other antitumor agents. For example, HDAC8 knockdown showed anti-proliferative activity in cells with a mutant, but not wild-type p53. Therefore, HDAC8 inhibitors might function as an adjuvant therapy for tumors with mutant p53.
These results are vividly demonstrated in the article entitled Discovery of meta-sulfamoyl N-hydroxybenzamides as HDAC8 selective inhibitors, recently published in the European Journal of Medicinal Chemistry. This work was conducted by Chunlong Zhao, Jie Zang, Qin’ge Ding, Wenfang Xu, and Yingjie Zhang from the School of Pharmaceutical Sciences, Shandong University, and Elizabeth S. Inks and C. James Chou from South Carolina College of Pharmacy, Medical University of South Carolina.
Related Posts
 Improving Tools For Quality Improvement: Crossings, Runs, And Crossrun
Improving Tools For Quality Improvement: Crossings, Runs, And Crossrun Coherent Poly Propagation Materials With 3-Dimensional Photonic Control Over Visible Light
Coherent Poly Propagation Materials With 3-Dimensional Photonic Control Over Visible Light Reconstructing Commuter Networks Using Machine Learning And Urban Indicators
Reconstructing Commuter Networks Using Machine Learning And Urban Indicators Surprising Internal Structure Of Whole Wheat-Green Gram Functional Bread
Surprising Internal Structure Of Whole Wheat-Green Gram Functional Bread Tailoring Tomatoes To Match Individual Consumer Needs
Tailoring Tomatoes To Match Individual Consumer Needs Eavesdropping On “Classroom Talk” In Undergraduate STEM Classrooms
Eavesdropping On “Classroom Talk” In Undergraduate STEM Classrooms