





It starts when the rain reaches the ground and percolates into the soil, infiltrating the first layers of the Earth’s crust where groundwater circulates. This water reacts with the rocks, dissolving some minerals and growing other ones.
In the presence of dissolved carbon dioxide, newly-formed minerals include various carbonate minerals such as calcite, magnesite, or siderite, which form by binding a dissolved carbonate group CO32- with calcium, magnesium, or iron, respectively. These minerals are widespread in the first kilometers of the Earth’s crust and may react with various other elements, including hazardous species such as arsenic, phosphate, selenium, or antimony. They can store these elements either as attached to the carbonate surface or directly incorporated in the crystalline structure.
Mineral-fluid reactions are usually modeled as occurring in the bulk fluid and following equilibrium geochemical theories. Such theories allow approximating the long-term evolution of complex fluid-rock systems that include various minerals, dissolved species, and water. However, at the atomic scale, the situation is more complex because several local out-of-equilibrium processes may control the formation of new minerals.
We have imaged the interactions between various carbonate minerals with either pure water or water that contained dissolved negatively-charged species (anions) such as arsenic, phosphate, selenium, and antimony. The technique of atomic force microscopy was employed because it allows imaging, as a function of time, the dissolution and precipitation processes over a mineral surface with nanometer height resolution and submicrometer spatial resolution.
In our experiments, we started with a given carbonate mineral and let some pure water flow over its surface, dissolving it. Dissolution occurs by the continuous removal of atomic layers from the carbonate. The dissolved species build a boundary layer of a thickness of a micrometer to a millimeter, where the fluid is locally more enriched than the bulk water into species coming from the dissolution of the carbonate. Such boundary layers act as microchemical reactors where new solid phases can form; the entire process is called an interface coupled dissolution-precipitation mechanism (Figure 1). When some toxic species are present in the fluid, they can be incorporated into these new solid phases, trapping them more permanently than if they kept dissolved in the fluid and transported with it.
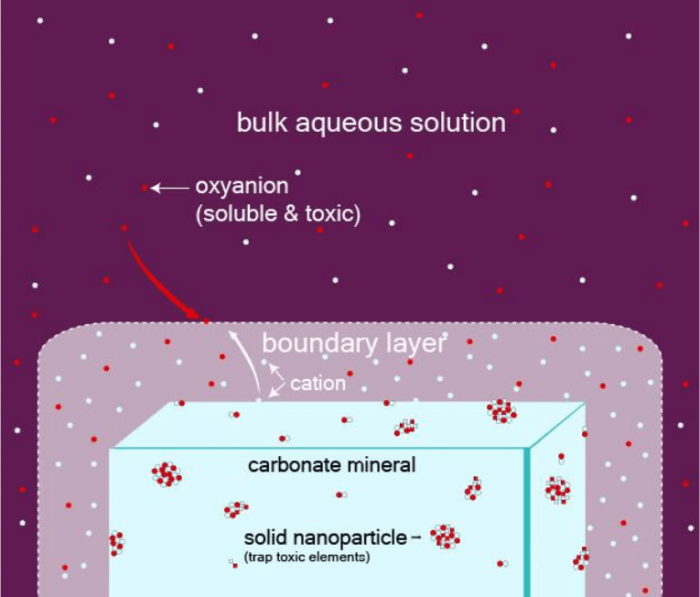
Figure 1: Concept of the coupled dissolution process that leads to the formation of new mineral nanoparticles at the surface of a carbonate grain. Because of the dissolution of the carbonate mineral, a boundary layer forms where new minerals can precipitate in the form of nanoparticles due to local out of equilibrium geochemical conditions. These nanoparticles may trap toxic and negatively charged (anion) elements initially present in the water. Figure courtesy François Renard.
We have studied several carbonate systems. When magnesite or dolomite, magnesium-bearing minerals, are the dissolving phase, nanoparticles of hydrated magnesium-rich minerals form at the surface. When calcite, a calcium-bearing carbonate, is the dissolving mineral, and in the presence of arsenic, phosphate, selenium, or antimony dissolved into the fluid, new minerals grow on the calcite surface in the form of nanoparticles (Figures 2, 3). When siderite, an iron-bearing carbonate, is the dissolving mineral, new precipitates are iron oxide minerals that can also trap some toxic species present in the fluid. This process of coupled dissolution-precipitation is therefore widespread, concerns most major carbonate minerals present in Earth surface environments, and can immobilize pollutants into solid phases.
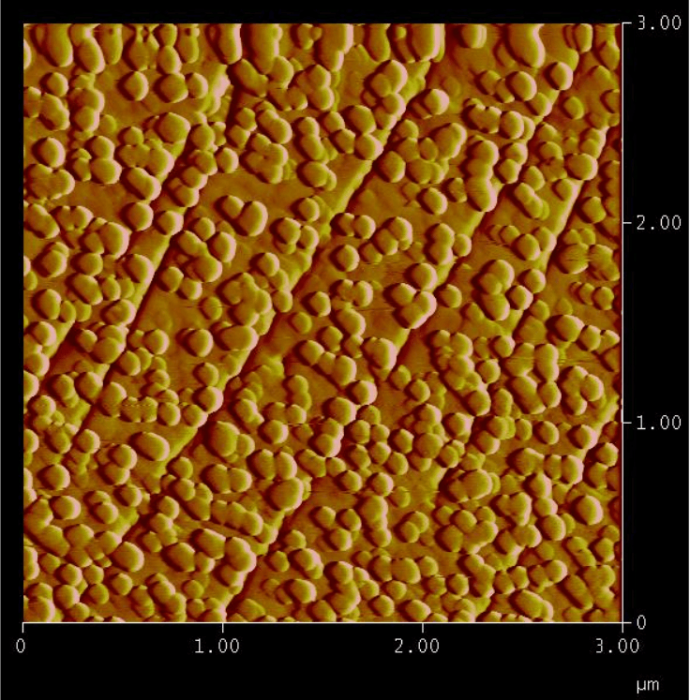
Figure 2: Trapping of arsenic in the form of circular nanoparticles that precipitate on a calcite surface during its dissolution at pH 1.6 in the presence of an arsenic-rich solution. Width of the picture: 3 micrometers. Height of the nanoparticles: 12 nanometers. Figure courtesy François Renard.
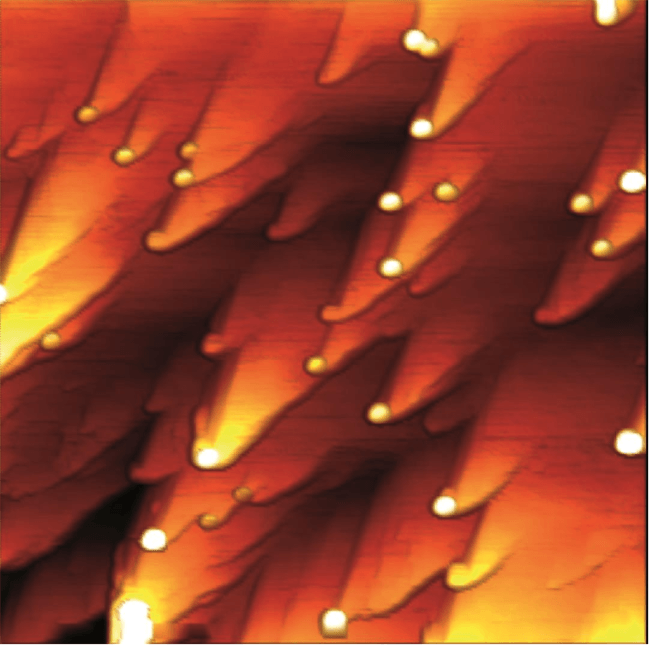
Figure 3: Trapping of antimony in the form of nanoparticles (circular white dots) attaching at the surface of a calcite mineral during its dissolution at pH 3 in the presence of and antimony-rich solution. Width of the picture: 5 micrometers. Height of the nanoparticles (white): 10 nanometers. Figure courtesy François Renard.
We have identified the parameters controlling the formation of nanoparticles. For a fluid at a given acidity (pH), the solubility of the carbonate mineral and its dissolution rate control the thickness of the boundary layer and the time scales of the formation of nanoparticles, as long as the precipitation of the nanoparticles is faster than the dissolution of the carbonate. The boundary layer thickness may vary from several micrometers to several millimeters. The time scales necessary for the formation of the first nanoparticles is in the range of seconds to hours. Our theoretical developments provide therefore predictions for the boundary layer thickness and for the time scale of nanoparticle formation that could be tested in other fluid-mineral systems.
The evolution of a mineral-fluid boundary layer is a dynamic process where dissolved species concentrations will vary depending on dissolution and precipitation rates. The interface-coupled process that occurs in a micro-reactor, the boundary layer, controls many transformations of carbonate minerals in shallow Earth environments. An important property of this process is that it happens locally, even if the fluid is undersaturated with respect to the new precipitates at the macroscopic scale. This phenomenon has important environmental implications for the trapping of pollutants on calcite surfaces because the mobility of these pollutants in circulating groundwater is reduced. Such an effect has implications not only for global geochemical cycles of those elements but also for the remediation of contaminated geochemical systems such as acid mine drainage and effluents of municipal waste landfills.
These findings are described in the article entitled Timescales of interface-coupled dissolution-precipitation reactions on carbonates, recently published in the journal Geoscience Frontiers.
Reference:
- Renard, F., Røyne, A., Putnis, C. V. (2019) Timescales of interface-coupled dissolution-precipitation reactions on carbonates, Geoscience Frontiers, 10, 17-27, https://linkinghub.elsevier.com/retrieve/pii/S1674987118300768.
Related Posts
 Duck-Billed Dinosaurs Uncovered In Aniakchak, Alaska
Duck-Billed Dinosaurs Uncovered In Aniakchak, Alaska Cryptic Diversity In Vietnam’s Limestone Karst Habitats
Cryptic Diversity In Vietnam’s Limestone Karst Habitats An Improved Method To Remove Debris From Cyst Nematode Egg Suspensions And Computer-Aided Technologies For Egg Counting
An Improved Method To Remove Debris From Cyst Nematode Egg Suspensions And Computer-Aided Technologies For Egg Counting The Footprints Of Urbanization, Industrialization, And Agriculture On River Beds: Heavy Metal Contamination Assessment And Source Identification In River Sediments In Eastern China
The Footprints Of Urbanization, Industrialization, And Agriculture On River Beds: Heavy Metal Contamination Assessment And Source Identification In River Sediments In Eastern China Aging Dolphins Via Pectoral Flipper Radiography
Aging Dolphins Via Pectoral Flipper Radiography Glycoalkaloids In Potatoes: The Effect Of Biostimulants And Herbicides
Glycoalkaloids In Potatoes: The Effect Of Biostimulants And Herbicides