





The aryl hydrocarbon receptor (AHR) was originally found in 1976 as the mediator of toxicity and induction of drug-metabolizing enzymes caused by the highly potent environmental contaminants, dioxins, and polycyclic aromatics. Subsequent studies disclosed AHR to be an over 600-million-year-old protein, whose homologs are critical to normal development in Drosophila (the common fruit fly) and to nervous system function in C. elegans (a model roundworm), although they do not bind dioxins. This triggered interest in exploring the possible physiological functions of AHR in mammals.
Generation of AHR knockout mice was a turning point in these attempts, crucially helping to establish that AHR is actually an important, or even key regulator of a number of mammalian cellular processes, including cell proliferation, differentiation, apoptosis, adhesion and migration; liver and blood vessel development; heart function; development and functioning of the male and female reproductive systems; and tumor formation.
A particularly hot focus in AHR research has been its critical role in the maintenance of intestinal mucosal integrity. AHR activity in type 3 innate lymphoid cells was shown to be indispensable for the normal development of intestinal lymphoid follicles and for stimulating mucosal epithelial cells to generate antimicrobial peptides. It also alleviates intestinal inflammation. Thereby, AHR regulates the composition of the intestinal microbiota. The relationship is bidirectional since intestinal microbes generate AHR activators. Recently, AHR has further been linked to body weight regulation because AHR deficiency protects mice from high-fat-diet-induced obesity and associated maladies.
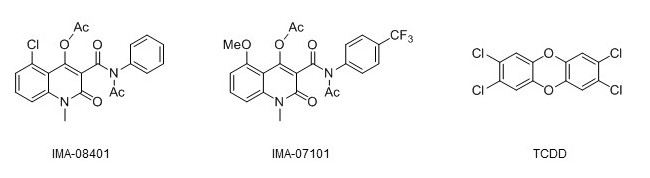
structural formulas of the two novel AHR agonists together with that of TCDD. Credit: Raimo Pohjanvirta
At the molecular level, AHR acts as a ligand-activated transcription factor, regulating the expression of a wide variety of genes. Among the most sensitive ones is CYP1A1 (involved in xenobiotic metabolism), whose gene expression or enzymatic activity is therefore commonly used as an index of AHR activity. In addition to this canonical pathway, AHR can act through other, alternative pathways, but their biological significance is still uncertain. Although no single substance has so far stood out as the “primary” endogenous ligand for the AHR, a number of diverse compounds have been shown to bind to it with varying avidities. By far the most important of these physiological agonists, such as 6-formylindolo(3,2-b)carbazole (FICZ) and kynurenine, are derivatives of tryptophan, an essential amino acid.
Due to the multifaceted physiological functions the AHR has proven to be involved in, interest has arisen to exploit it in treatments of various diseases. However, this approach will require great care to find or design such compounds that are potent AHR modulators but do not bring about the dioxin-like toxicity. We recently demonstrated that two such compounds, IMA-08401 and IMA-07101, are far less deleterious than the most toxic dioxin, TCDD, in rats. In the present study, we assessed the toxicity and potency of their active metabolites (which are rapidly generated by the parent compounds in mammals) in cell culture and modeled their binding to the AHR. Both showed little cytotoxicity to H4IIE rat hepatoma cells or bacterial mutagenicity in the Ames test. However, one of them slightly increased the number of micronuclei in H4IIE cells, suggesting a genotoxic effect. Although this finding warrants further studies, it is probably of minor significance because TCDD (known to be devoid of direct genotoxicity) exhibited a similar tendency.
As inducers of CYP1A1 activity, both compounds displayed potency comparable to TCDD but a shorter effect duration. This was in line with our previous findings in rats. Interestingly, a computerized simulation of ligand binding by molecular docking to a homology model of the AHR ligand-binding domain (a central part of the protein at which it is in contact with the ligand) revealed that these novel AHR agonists interact with the same amino acid residues in this region as TCDD. Hence, the rate of metabolism probably plays a major part in the case of differences in toxicity between TCDD and IMA-08401 & IMA-07101, whereas ligand binding is of low importance in this regard.
Thanks to their apparent low toxicity and high potency as AHR agonists, these two novel selective AHR modulators are promising candidates for therapeutic uses.
These findings are described in the article entitled In vitro toxicity and in silico docking analysis of two novel selective AH-receptor modulators, recently published in the journal Toxicology in Vitro.
Related Posts
 Important Strategies To Help Healthcare Providers Support Patients With Diabetes
Important Strategies To Help Healthcare Providers Support Patients With Diabetes Studying The Link Between Increased BMI And Late-Onset Preeclampsia In Pregnant Women
Studying The Link Between Increased BMI And Late-Onset Preeclampsia In Pregnant Women A Bacterial Cell Imaging Method Using CRISPR And Microfluidics
A Bacterial Cell Imaging Method Using CRISPR And Microfluidics B. infantis Reduces Key Markers Of Intestinal Inflammation In Infants
B. infantis Reduces Key Markers Of Intestinal Inflammation In Infants Epigenetic Changes In Multiple Sclerosis – Studied In Twins
Epigenetic Changes In Multiple Sclerosis – Studied In Twins Did You Know Math Can Help Us Learn How Diseases Work?
Did You Know Math Can Help Us Learn How Diseases Work?