





The atmospheric level of CO2 is rising dramatically more than ever in the Earth’s history, with projections that it could reach nearly 600 ppm by 2100. The excessive anthropogenic (or industrial) emissions of CO2, mostly originating from fossil-based systems, lead to irreversible disruptive impacts on climate and environment, such as global warming, glacial ablation, ocean acidification, rising of sea level, and extinction of species.
To alleviate these adverse effects, governments have inked the Paris Agreement, attempting to reduce atmospheric CO2 concentration. However, in 2016, MIT concluded that the goal provided by the Paris Agreement could not be met in the future under the current circumstances using their Integrated Global System Modeling (IGSM) to predict temperature increase results in 2100. In other words, only a combination of both emission reduction and removing CO2 from the atmosphere can produce enough of an effect. So both CO2 capture and utilization is needed.
CO2 can be converted via diverse ways, such as thermochemical, electrochemical, photochemical, radiochemical, and biochemical reactions. Of these methods, electrocatalytic CO2 reduction is especially appealing in term of minimizing environmental impact, given compatibility of the process with renewable energy systems. This process allows for sustainable and low carbon-footprint chemistry. There are many benefits associated with this process: 1) the reaction can be conducted at room temperature and pressure and in neutral pH; 2) it can proceed without feeding H2, but where the H2 is generated in situ by water electrolysis; 3) it converts intermittent electricity into stable chemical energy and generates valuable products; and 4) the product distribution can be tuned by adjusting external parameters (such as electrocatalysts, electrolytes, reaction temperature, and applied voltages).
Converting CO2 into fuels or commodity chemicals ushers in hope for an energy transition from today’s “fossil fuel economy” to a sustainable “CO2 economy.” The final products are up to twenty including carbon monoxide, formate, methane, ethane, ethylene, ethanol, and so on. Relative to C1 products, C2+ (containing two or more carbon atoms) hydrocarbons and oxygenates have higher energy densities. Also, many multi-carbon products are chemical feedstocks in high demand.
However, there remains a challenge for efficient CO2 reduction to C2+ species owing to 1) the competitive undesired H2O reduction; 2) large C-C coupling activation barrier as well as competition of C-C bond formation with C-O and C-H bond formations; and 3) overpotential gap between the formation of necessary CO intermediates and that of C2+ species. The adsorption energies of reaction intermediates seem to follow linear scaling relationships. The catalytic surface needs to bind *CO intermediates strongly to enable a sufficient coverage for further reduction or C-C coupling, whereas the associated activation barriers also increase with stronger *CO binding. In these scenarios, developing catalysts with appropriate electronic properties becomes critically important to optimize the interplay between the two criteria for selective C-C coupling.
By transferring a concerted proton-electron (H+/e–) from solution to adsorbed species, a CO2 molecule is reduced to a carboxyl intermediate *COOH and then to *CO and a water molecule. *CO is proposed to be a common intermediate for the production of different products. For C2+ products, from *CO intermediate there are two different reaction mechanisms.
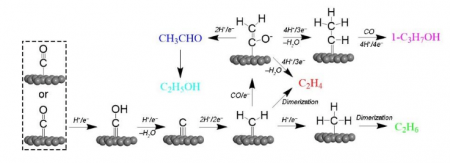
Fig. 1 Possible reaction pathways via coupling of two *CH2 species or CO insertion in a Fischer-Tropsch-like step. This image is provided by Zhenyu Sun.
In a “carbene” mechanism, *CO combined with proton and electron converting into *C, which can be transformed to *CH2. C2+ products are generated through either nonelectrochemical coupling of *CH2 moieties or a Fischer-Tropsch-like combination of *CH2 and CO (CO insertion), as shown in Fig. 1.
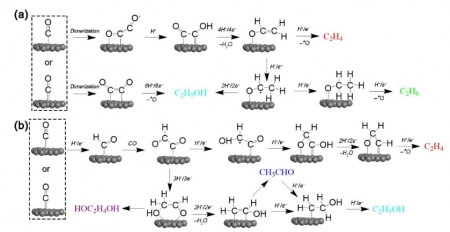
Fig. 2 Possible reaction pathways via (a) *CO dimerization and (b) *CHO step. This image is provided by Zhenyu Sun.
Other major routes reported for C2+ product generation involve *CO dimerization, a key step for C-C coupling at low overpotentials. *CO dimerizes to form *C2O2– (a rate-limiting step) and is then rapidly hydrated to *CO-COH via the Eley-Rideal (ER) mechanism using H2O + e– (H2Oad) (Fig. 2a). *CO-COH is reduced to vinyl alcohol (*CH2=CHO), a selectivity-determining intermediate. Subsequently, the pathway bifurcates into ethylene-forming, ethane-forming, and ethanol-forming routes. Sometimes, *CHO is predicted to be more stable than *C2O2-. This makes the reduction of *CO to *CHO which then reacts with *CO to form *COCHO more favorable over *CO dimerization and subsequent reduction (Fig. 2b). The *COCHO is key in determining selectivity between ethylene and ethanol. The formation of *COCHOH leads to ethylene, while *CH2CHOH forms ethanol.
To promote the formation of C2+ compounds over C1 products and H2, we need to make attempts to manipulate adsorption energy and surface coverage of C2+ intermediates. Maximization and stabilization (optimization of binding energies) of key reaction intermediates such as *CO, *COH, *COCO should be enhanced via surface modification, structure tuning, and creation of tandem systems with synergistic/cooperative multiple active sites. Inspired by enzyme catalysis, construction of three-dimensional (3D) active sites likely break the scaling relation limitation. Distinct active centers in close proximity in a 3D porous space can bind different sites of intermediates, thereby facilitating C2+ product generation.
These discussions and summarizations are provided in the article entitled Electrochemical CO2 reduction to C2+ species: Heterogeneous electrocatalysts, reaction pathways, and optimization strategies, recently published in Materials Today Energy. This work was conducted by Qun Fan, Mingli Zhang, Mingwen Jia, Shizhen Liu, Jieshan Qiu, and Zhenyu Sun from State Key Laboratory of Organic-Inorganic Composites, College of Chemical Engineering, Beijing University of Chemical Technology.
Related Posts
 Be Careful When Speaking About Lead Pollution: The Good, The Bad, And The Ugly!
Be Careful When Speaking About Lead Pollution: The Good, The Bad, And The Ugly! Temperature Has A Significant Influence On The Production Of SMP-Based Dissolved Organic Nitrogen (DON) During Biological Processes
Temperature Has A Significant Influence On The Production Of SMP-Based Dissolved Organic Nitrogen (DON) During Biological Processes Does The Arrow Of Time Apply To Quantum Systems?
Does The Arrow Of Time Apply To Quantum Systems? Labeled Periodic Table
Labeled Periodic Table A “Twist” In Wavefunction With Ultrafast Vortex Electron Beams
A “Twist” In Wavefunction With Ultrafast Vortex Electron Beams Chemical And Biological Characterization Spot The Faith Of Nanoparticles
Chemical And Biological Characterization Spot The Faith Of Nanoparticles