





Most of the energy needed to sustain economic growth, since the industrial revolution, is derived from fossil sources: natural gas, coal, and oil. In utilizing these sources in industry, the first process and probably the most important is that of the so-called “reforming” of hydrocarbons. The process at which water in the form of steam reacts with natural gas (mostly methane) at high temperatures producing carbon monoxide and hydrogen (syngas) from which a large fraction of chemicals are produced. At the heart of all these processes is the catalyst; often in the form of a metal dispersed on an oxide support in mono- or multiphase form.
The main carrier of energy in these chemical processes is hydrogen, thus making it is at the essence of petrochemical industry. Yet, its origin from methane by steam reforming makes the process among the most polluting on earth. For each kg of hydrogen, stoichiometrically, 5.5 kg of CO2 are released (not taking into account the high-energy cost of the process). CO2 is a harmful greenhouse gas and therefore contributes to the steady rise of the earth’s atmospheric temperature. Great efforts are being made at present to convert CO2 to other products. Any process involving CO2 conversion is energy intensive, climbing the thermodynamic ladder. Therefore, CO2 conversion to other products can only contribute positively into the environment if the needed electrons to reduce it are provided from renewable sources. The most abundant chemical on earth in liquid form is water and it is, therefore, the cheapest possible source for hydrogen, provided that the electrons needed to reduce the two hydrogen ions of water originate from a universally stable source such as the sun in this case.
Water can be split into molecular hydrogen and oxygen via many methods such as electrolysis and photocatalysis; however, the efficiency of the latter is far lower than that of the former. This is because a photocatalyst, at the nanoscale, consists of a solar cell (a multiple stacks of semiconductors and metals) and an electrolyzer (often composed of noble metals for hydrogen ion reduction and a metal oxide for oxygen anion oxidation). The process is, therefore, very complex, probably at present escaping human knowledge. As of today, efficient photocatalysts are not stable, and stable photocatalysts are of anecdotal efficiency. The reasons for that are multiple and mostly relay on light-matter interaction at the nanoscale, thermodynamics dictating band edges alignment of the involved semiconductor, and the complex kinetics of the reaction steps.
The Field of Photocatalysis and Its Potential
In photocatalysis, a catalyst uses photons to excite its electrons from highest occupied electronic states (valence bands) to lowest unoccupied ones (conduction band). This allows for a redox reaction to occur if the band energies are well tuned with their potentials. Based on the solar light spectrum reaching the earth’s surface, the theoretical maximum of a solar cell is approximately 34% and referred to as the Shockley–Queisser limit. [1]
What makes the concept of photocatalysis attractive is the potential process simplicity in the chemical industry along with low operation cost in the reaction section if the catalyst is sufficiently efficient. This is the main motivation behind investigating the fundamental aspects of chemical reactions and materials properties to develop an efficient and stable photocatalyst. This is in particular important because of the low density of light hitting the surface of the earth making the use of very large areas needed which in turn makes the process very sensitive to the overall efficiency. Researchers in the field thus utilize many tools such as computational simulations to study properties of potential and/or prototype photocatalysts in terms of electronics, geometrical, optical properties in addition to surface interaction with reactive molecules to ultimately design better photocatalysts.
Computational Tools as Guiding Scopes
Many materials pose a potential to be an efficient photocatalyst based on their energy band gap and the available solar light, nevertheless, most prove to be inefficient when tested. Investigating a basic reaction such as water splitting may seem trivial but in reality is very complex. This is in general due to multiple pathways the reaction can follow and the not-well-understood interaction between reactants and photocatalyst. [2] By simulating a surface of a photocatalyst, one can estimate the material properties and its interaction with the molecules of interest.
Titania (TiO2) is an abundant oxide with a bandgap that theoretically can absorb up to 4-5% of the solar spectrum but is inefficient in reality in performing total water splitting. Since TiO2 is considered a prototype for many photocatalytic reactions, it is under fundamental investigation by researchers in the field; mostly due to its stability. Organic molecules such as ethanol can be easily oxidized by excited TiO2 in contrast to water, and are thus used as prototype molecules for comparative investigations. During the process of water oxidation, H2O2 as a reaction intermediate has been postulated and, therefore, its transformation to O2 and H2 can be a limiting step.
2 H2O (liquid) H2(gas) + H2O2(adsorbed intermediate) ΔG= + 354 kJ/mol
H2O2(adsorbed intermediate) H2(gas) + O2(gas) ΔG= + 120 kJ/mol
We have built a slab of TiO2 to study its properties along with the reactant molecule and its constituents using Density Functional Theory (DFT) [3]. One of the most valuable information obtained by such model is the adsorption energy that unveils how strong does a molecule or its intermediates stick to the surface of the slab to ultimately allow a reaction to happen (via electron transfer). The adsorption energy can also be supplemented by studying the final geometrical coordinates of simulated atoms to calculate, for example, bond lengths and angles. This can be performed by allowing the simulated atoms to relax by finding the coordinates that give the lowest energy to the total system.
Our results showed that both ethanol and hydrogen peroxide in their molecular and dissociative forms have relatively high adsorption energies (0.8-1.1 eV). This indicates that all are theoretically stable on the surface and conceptually reactive. However, looking closely into the electronic structure represented by Density of States (DOS), we concluded that ethanol is reactive and H2O2 is not due to the missing electronic orbital overlap required for electron transfer in the case of H2O2 and its constituents. The orbitals under investigation are the two highest occupied molecular orbitals (HOMO) of the adsorbates and the orbitals of the surface oxygen of the photocatalyst as presented in Figure 2. Ethanol and ethoxide (Figure 2A-B) both have pronounced overlap in contrast to H2O2 in its molecular and dissociative forms (Figure 2C-E) where negligible overlap exists.
In conclusion, our investigation indicated that the absence of a complete reaction of water on TiO2 to molecular hydrogen and oxygen can be linked to the formation of a “relatively” stable H2O2 molecule on the surface hindering the reaction.
These findings are described in the article entitled Study of the modes of adsorption and electronic structure of hydrogen peroxide and ethanol over TiO2 rutile (110) surface within the context of water splitting, recently published in the journal Surface Science. This work was conducted by H. Alghamdi and H. Idriss from SABIC Corporate Research and Development.
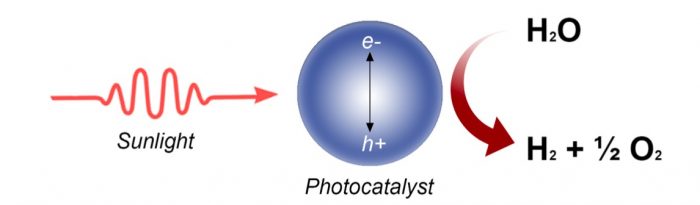
Figure 1 Schematic representing how a photocatalyst works to split water. Credit: Hamdan Alghamdi
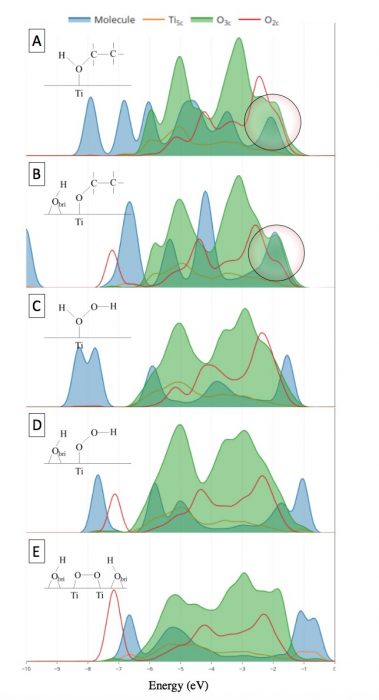
Figure 2 Projected density of states of (A) ethanol, (B) ethoxide, (C) H2O2, (D) HOO, and (E) OO on TiO2 rutile (110) surface. Credit: Hamdan Alghamdi
References:
- Shockley, William, and Hans J. Queisser. “Detailed Balance Limit of Efficiency of p‐n Junction Solar Cells.” Journal of Applied Physics, vol. 32, no. 3, 1961, pp. 510–519., doi:10.1063/1.1736034.
- Henderson, M. A., The interaction of water with solid surfaces: fundamental aspects revisited. Surface Science Reports, vol. 46, 2002, pp. 1-308., doi.org/10.1016/S0167-5729(01)00020-6
- Alghamdi, H., and H. Idriss. “Study of the Modes of Adsorption and Electronic Structure of Hydrogen Peroxide and Ethanol over TiO2 Rutile (110) Surface within the Context of Water Splitting.” Surface Science, vol. 669, 2018, pp. 103–113., doi:10.1016/j.susc.2017.09.011.
Related Posts
 Be Careful When Speaking About Lead Pollution: The Good, The Bad, And The Ugly!
Be Careful When Speaking About Lead Pollution: The Good, The Bad, And The Ugly! Temperature Has A Significant Influence On The Production Of SMP-Based Dissolved Organic Nitrogen (DON) During Biological Processes
Temperature Has A Significant Influence On The Production Of SMP-Based Dissolved Organic Nitrogen (DON) During Biological Processes Does The Arrow Of Time Apply To Quantum Systems?
Does The Arrow Of Time Apply To Quantum Systems? Labeled Periodic Table
Labeled Periodic Table A “Twist” In Wavefunction With Ultrafast Vortex Electron Beams
A “Twist” In Wavefunction With Ultrafast Vortex Electron Beams Chemical And Biological Characterization Spot The Faith Of Nanoparticles
Chemical And Biological Characterization Spot The Faith Of Nanoparticles