





Nuclear energy has immense potential because uranium and other actinides are the most energy-dense resources available. For instance, only a few grams of uranium dioxide releases the same amount of energy in a nuclear reactor that is produced at a burning of several hundred kilograms of anthracite coal or oil. Under operating conditions, the fuel in the pellets undergoes complex phase and structure transitions caused by temperature and radiation impacts.
Currently, there is a challenge in nuclear materials science dealing with the accurate description (and prediction) of nuclear fuel behavior under operating conditions. Several methods may resolve this task, but the most perspective approach is the multiscale computer modeling. It implies that various techniques of modeling and theoretical physics are used for the solution of subtasks on various time and spatial scales. The cooperation of different computational approaches allows predicting behavior of the nuclear materials under different conditions. Such modeling can reduce the number of experiments that are necessary at a final stage of fuel verification. It is especially crucial for a new type of nuclear reactors, where the experiments on the fuel degradation can last up to a dozen years.
There is a variety of nuclear fuels (namely, UO2, UN, UC, metallic fuels), and each of them has its unique thermophysical properties. In our recent study, we investigate the properties of point defects in the fuel crystal lattice. Our study is based on the combination of the atomistic simulation with the thermodynamic approach to the description of defects concentration. We showed that some types of phase transitions in the fuel may be described as the accumulation of point defects. The experimental data about similar phenomena is minimal. For instance, there is no information about the width of homogeneity area on the phase diagram for uranium mononitride UN. This area was only known to be extremely narrow and much smaller than that for uranium dioxide or uranium monocarbide.
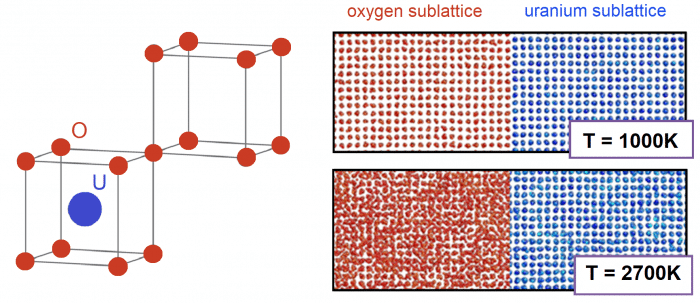
The crystal structure of uranium dioxide (left figure) and the snapshots of the atomistic simulations at heating of UO2 (right figures). The simulation fragments correspond to the low temperature before the superionic transition (T= 1000 K) and the high temperature after the superionic transition (T = 2700 K). For convenience, the oxygen and uranium sublattices are shown separately. Credit: Sergei Starikov
In our work, we have calculated the permissible variation of UN composition near stoichiometric component ratio (1:1) that allows conserving a fuel homogeneity. The main reason for the extremely narrow homogeneity area for UN is an unusual energy hierarchy of point defects. At the stoichiometric conditions, the concentrations of almost all point defects are comparable to each other. Relatively small deviations of the composition from stoichiometric state lead to a saturation of the uranium sublattice by the point defects. The limiting concentrations of these defects cause the formation of new phases.
Another interesting phenomenon studied in our work is the transition to a superionic state in UO2. Such processes take place in binary systems, when one of the sublattices accumulates defects, while the other one remains defectless. Such defects accumulation takes place during heating and continues until almost full disordering of one of the sublattices. Sometimes this process is called premelting. However, in contrast to the true melting, it has the attributes of a second-order phase transition (i.e., continuous change in volume and energy with a change in temperature). For uranium dioxide, the discussion about the existence of this transition is under consideration since the middle of the 20th century. Transition temperature for this fuel is about 2500 K, and such high temperature is the main reason for enduring discussions about details of the process. Our atomistic simulation allows studying the properties of the superionic transition. In particular, we show that the superionic transition temperature can be estimated only from the formation energy of point defects.
The developed models and simulation results obtained in the present work can be implemented in the macroscale computational codes describing the evolution of nuclear fuel properties at operating conditions. Such codes are the necessary tools for nuclear engineering since they allow to significantly increase the accuracy of forecasts about nuclear material behavior and, hence, provide better control.
These findings are described in the article entitled Description of phase transitions through accumulation of point defects: UN, UO2 and UC, recently published in the Journal of Nuclear Materials. This work was conducted by S. Starikov and M. Korneva from the Joint Institute for High Temperatures of the Russian Academy of Sciences.
This work is a continuation of the research on the description of the nuclear fuel properties based on atomistic simulation, reported previously in [A. Lunev, S. Starikov et al. Understanding thermally-activated glide of screw dislocations in UO2 – A molecular dynamics analysis, International Journal of Plasticity (2018)] and in [A. Kuksin, S. Starikov et al. The diffusion of point defects in uranium mononitride: Combination of DFT and atomistic simulation with novel potential, Journal of Alloys and Compounds (2016)]
Related Posts
 Improving Tools For Quality Improvement: Crossings, Runs, And Crossrun
Improving Tools For Quality Improvement: Crossings, Runs, And Crossrun Coherent Poly Propagation Materials With 3-Dimensional Photonic Control Over Visible Light
Coherent Poly Propagation Materials With 3-Dimensional Photonic Control Over Visible Light Reconstructing Commuter Networks Using Machine Learning And Urban Indicators
Reconstructing Commuter Networks Using Machine Learning And Urban Indicators Surprising Internal Structure Of Whole Wheat-Green Gram Functional Bread
Surprising Internal Structure Of Whole Wheat-Green Gram Functional Bread Tailoring Tomatoes To Match Individual Consumer Needs
Tailoring Tomatoes To Match Individual Consumer Needs Eavesdropping On “Classroom Talk” In Undergraduate STEM Classrooms
Eavesdropping On “Classroom Talk” In Undergraduate STEM Classrooms