





S. aureus is a type of bacterium that is carried in the nose of 30% of healthy adults and can cause diseases ranging from mild to severe. It is categorized as a serious-threat pathogen, especially because of its low susceptibility to many classes of antibiotics.
Efforts to find new antibiotic regimens have shown that the germ’s environment affects the efficacy of antibiotics, hinting that multiple mechanisms could be in play. Rising evidence demonstrates that there is a clear link between antibiotic susceptibility and metabolism, with a trade-off between the two. For example, bacteria with low metabolic activity are less susceptible to a subset of antibiotics. In other words, antibiotic resistance is a systems-level phenomenon.
Reconstructed metabolic networks exist for many bacteria including S. aureus. They include information about metabolites, chemical transformations, and participating metabolic genes. When they are converted into models, multiple methods can be used to compute metabolic activity across the network in different conditions. To get reliable results, the metabolic networks are curated in multiple iterations and updated as new discoveries are made. Curations involve the addition of novel information and removal of erroneous content, as well as the adjustment of thermodynamic constraints.
In our recent paper published in Plos Computational Biology, we curated a network of metabolism specific to S. aureus. By pooling together from the literature all of the recently discovered metabolic processes into pathways, we achieved an increase of 40% in content. In addition, we enriched the network with three-dimensional protein structures mapping to the modeled metabolic genes, a new feature of metabolic networks that has been used to analyze mutations on the network level.
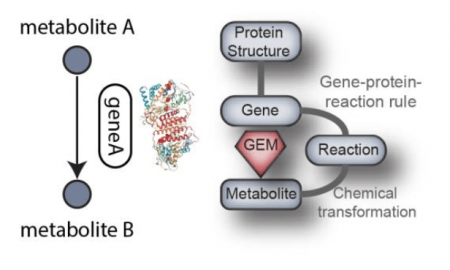
Credit: Yara Seif
Flux balance analysis is a popular method to compute metabolic activity across the network. In its most common form, it simulates the activity across the network that achieves the maximum production of biomass components (such as cell wall, DNA, RNA, protein, etc.) which are often end products of metabolism. Using flux balance analysis, we predicted the effect of removing each modeled gene (and the corresponding metabolic reaction(s)) on biomass production. We compared our results to experimental observations (in which the effect of knocking out of a gene is measured by the bacteria’s ability to grow) and obtained 85% agreement. We repeated our simulations by modeling the nutritional landscape in the nose and blood and found that nucleotide and amino acid biosynthesis are essential in the two environments.
We also evaluated the effect of glucose on the intracellular metabolic activity levels using the measured concentration of extracellular metabolites across time in two conditions (with and without glucose). We found that there was increased activity in branched-chain amino acid biosynthesis, glycolysis, and the pentose phosphate pathway in the presence of glucose, with overflow pathways contributing to the production of ATP, and the pentose phosphate pathway contributing to the NADPH pool. In the absence of glucose in the medium, the Krebs cycle, gluconeogenesis, and amino acid utilization were more prevalent with the NADPH pool being partially recycled as a by-product of the Krebs cycle, and ATP being predominantly produced in the respiratory pathway.
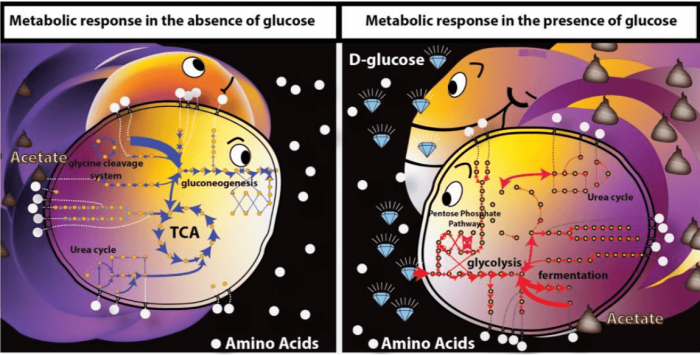
Credit: Yara Seif
The genome-scale metabolic model reconstructed in this work can be utilized as a common platform and constitutes a stepping stone toward understanding the systems-level metabolic response of S. aureus under various conditions.
These findings are described in the article entitled A computational knowledge-base elucidates the response of Staphylococcus aureus to different media types, recently published in the journal PLOS Computational Biology.
Related Posts
 Important Strategies To Help Healthcare Providers Support Patients With Diabetes
Important Strategies To Help Healthcare Providers Support Patients With Diabetes Studying The Link Between Increased BMI And Late-Onset Preeclampsia In Pregnant Women
Studying The Link Between Increased BMI And Late-Onset Preeclampsia In Pregnant Women A Bacterial Cell Imaging Method Using CRISPR And Microfluidics
A Bacterial Cell Imaging Method Using CRISPR And Microfluidics B. infantis Reduces Key Markers Of Intestinal Inflammation In Infants
B. infantis Reduces Key Markers Of Intestinal Inflammation In Infants Epigenetic Changes In Multiple Sclerosis – Studied In Twins
Epigenetic Changes In Multiple Sclerosis – Studied In Twins Did You Know Math Can Help Us Learn How Diseases Work?
Did You Know Math Can Help Us Learn How Diseases Work?