





Neurodevelopmental and neuropsychiatric disorders such as autism spectrum disorder (ASD) and schizophrenia are known to affect about 1% of the general population. Genetic factors are believed to make a substantial contribution. However, though massive genome sequencing efforts in recent years have uncovered a number of genes associated with these disorders, the underlying cause remains largely unknown for the majority of patients.
Fortunately, there is also a relatively small group of ASD patients for whom we know very well what the genetic causes are, and a great deal of what we currently know about ASD has come from studying what is commonly known as “syndromic” forms of ASD. Syndromic ASD is a group of genetic disorders with clinically defined criteria which includes fragile X syndrome, neurofibromatosis, tuberous sclerosis, Rett syndrome, and Angelman syndrome, just to name a few. The causal genes for these syndromic forms of ASD have been identified as FMRP, NF1, TSC1/TSC2, MECP2, and UBE3A, respectively. With the causal gene known, scientists have been able to study these disorders by creating animal models by removing the specific gene or creating similar mutations in them as in human patients.
Interestingly, many of the causal genes of syndromic ASD center upon a particular signaling pathway in the cell called mTOR (mechanistic target of rapamycin). The loss of function or mutations in the causal genes often result in excessive activation of mTOR signaling, causing havoc on the various cellular process that it controls. Importantly, one of the processes that mTOR inhibits is a degradative mechanism called autophagy, which equips the cell with the ability to break down unwanted or damaged cellular components to their basic parts so that they could be reused again. There have been several studies in recent years to suggest that perhaps some ASD patients have lower autophagic activity in their brains, and that mice without autophagy in the brain exhibit ASD-like abnormalities. However, it was never clearly understood why autophagy would be relevant to neurodevelopmental and neuropsychiatric disorders.
In our recent study, we specifically removed a gene, Atg7, which is important for the initiation of autophagy, from two different types of brain cells that are believed to be important in neurodevelopmental and neuropsychiatric disorders: excitatory and GABAergic inhibitory neurons. When we tested the mutant animals for behavioral abnormalities, we found that they both showed similar signs of reduced social interaction and increased anxiety and distress. Because autophagy is important for cleaning up the unwanted materials in the cell, brain cells without autophagy accumulate protein aggregates — clumps of proteins that are not in the right place or shape to carry out their usual functions. We hypothesized that whatever was causing the animals to behave abnormally is likely due to this buildup of unwanted materials in the cell, so we devised a new way to identify what those aggregated proteins are by quantitative mass spectrometry.
One of the proteins we found by this approach is called GABARAPL2, or γ-aminobutyric acid (GABA) receptor-associated, protein-like 2, which belong to a group of proteins that are believed to help bring GABAA receptors to the cell surface. GABA is the main inhibitory neurotransmitter in the brain, therefore making GABA signaling very important for effective communication between different brain cells and different parts of the brain. To our surprise, we found that this group of proteins, collectively called GABARAPs, were trapped in the protein aggregates that normally form in cells that have no or reduced autophagic activity. Furthermore, not only were they in the wrong place, we also found that they were not in their usual conformations and therefore probably could no longer function properly.
We tested this idea by examining the surface levels of GABAA receptors and indeed found that mutant brain cells had fewer of these receptors exposed, consistent with the trafficking functions of GABARAPs being compromised in mice without autophagy in their brains. We further performed electrophysiology experiments to help confirm that inhibitory inputs into affected brain cells were reduced and that they had, in turn, became more active.
In order to probe further into how the protein aggregates, formed as a result of autophagy disruption, affected GABA signaling, we artificially made normal wild-type brain cells to produce a large amount of a protein called p62, a key component of the protein aggregates formed in the absence of autophagy. We found that the excess p62 also formed aggregates that trapped GABARAPs and that the movement of GABAA receptors to the cell surface was also reduced. Conversely, we observed that it was possible to reverse the change in surface GABAA receptor by reducing p62 expression using small hairpin RNA in autophagy-deficient brain cells, demonstrating that the main culprit was the p62+ protein aggregates. Perhaps more importantly, we also found that a subpopulation of ASD patients had increased aggregation of GABARAPs and p62 in their brains. Interestingly, these patients also tended to have less TSC2 proteins in their brains, suggesting that there was likely excessive mTOR signaling, which in turn suppressed autophagy and caused GABARAPs and p62 to accumulate and aggregate.
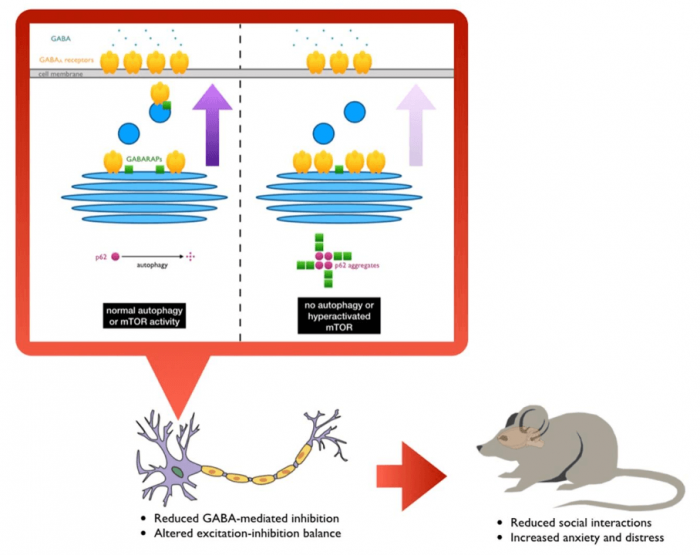
Figure courtesy Kelvin Hui and RIKEN.
Collectively, our study demonstrated that mTOR-regulated autophagy makes a substantial contribution to ASD pathology by tipping the balance between excitatory and inhibitory neurotransmission. Furthermore, since recent studies have also implicated mTOR dysfunction in other neurodevelopmental and neuropsychiatric disorders, the molecular mechanism that we have uncovered in this study may also be involved in those disorders.
Our findings also highlight the importance of protein homeostasis in the brain for its proper functioning. Unwanted accumulation of proteinaceous materials may, in fact, have wider consequences as they attract and trap otherwise functional proteins into them. This is analogous to our recent studies in which the co-aggregation of risk factor proteins of neuropsychiatric disorders, DISC1 for example, with protein aggregates formed in neurodegenerative diseases such as Huntington’s disease and frontotemporal dementia may underlie psychiatric symptoms often observed in patients with neurodegenerative diseases.
This was the first demonstration of a link between mTOR and GABA signaling, which separately have been under intense research focus in the area of neurodevelopmental and neuropsychiatric disorders. Even though these findings were made within the context of the brain, we wonder whether this newly identified link may also apply to other disease conditions in which both mTOR and GABA signaling have been implicated, such as cancer and diabetes. Under such circumstances, one may expect that novel drugs that can induce autophagy to help with maintaining protein homeostasis or directly disrupt the co-aggregation of p62 and GABARAPs to provide the desired therapeutic effects to a wide range of human pathologies.
These findings are described in the article entitled GABARAPs dysfunction by autophagy deficiency in adolescent brain impairs GABAA receptor trafficking and social behavior, recently published in the journal Science Advances.
References:
- Costa-Mattioli, M., and Monteggia, L.M. (2013). mTOR complexes in neurodevelopmental and neuropsychiatric disorders. Nature Neuroscience 16, 1537–1543.
- Endo, R., Takashima, N., Nekooki-Machida, Y., Komi, Y., Hui, K.K.-W., Takao, M., Akatsu, H., Murayama, S., Sawa, A., and Tanaka, M. (2018). TAR DNA-Binding Protein 43 and Disrupted in Schizophrenia 1 Coaggregation Disrupts Dendritic Local Translation and Mental Function in Frontotemporal Lobar Degeneration. Biological Psychiatry 84, 509–521.
- Hui, K.K., Takashima, N., Watanabe, A., Chater, T.E., Matsukawa, H., Nekooki-Machida, Y., Nilsson, P., Endo, R., Goda, Y., Saido, T.C., et al. (2019). GABARAPs dysfunction by autophagy deficiency in adolescent brain impairs GABAA receptor trafficking and social behavior. Sci Adv 5, eaau8237.
- Hui, K., Katayama, Y., Nakayama, K.I., Nomura, J., and Sakurai, T. (2018). Characterizing vulnerable brain areas and circuits in mouse models of autism: Towards understanding pathogenesis and new therapeutic approaches. Neuroscience & Biobehavioral Reviews.
- Leil, T.A., Chen, Z.-W., Chang, C.-S.S., and Olsen, R.W. (2004). GABAA receptor-associated protein traffics GABAA receptors to the plasma membrane in neurons. J. Neurosci. 24, 11429–11438.
- Tanaka, M., Ishizuka, K., Nekooki-Machida, Y., Endo, R., Takashima, N., Sasaki, H., Komi, Y., Gathercole, A., Huston, E., Ishii, K., et al. (2017). Aggregation of scaffolding protein DISC1 dysregulates phosphodiesterase 4 in Huntington’s disease. J Clin Invest 127, 1438–1450.
- Tang, G., Gudsnuk, K., Kuo, S.-H., Cotrina, M.L., Rosoklija, G., Sosunov, A., Sonders, M.S., Kanter, E., Castagna, C., Yamamoto, A., et al. (2014). Loss of mTOR-dependent macroautophagy causes autistic-like synaptic pruning deficits. Neuron 83, 1131–1143.
Related Posts
 Important Strategies To Help Healthcare Providers Support Patients With Diabetes
Important Strategies To Help Healthcare Providers Support Patients With Diabetes Studying The Link Between Increased BMI And Late-Onset Preeclampsia In Pregnant Women
Studying The Link Between Increased BMI And Late-Onset Preeclampsia In Pregnant Women A Bacterial Cell Imaging Method Using CRISPR And Microfluidics
A Bacterial Cell Imaging Method Using CRISPR And Microfluidics B. infantis Reduces Key Markers Of Intestinal Inflammation In Infants
B. infantis Reduces Key Markers Of Intestinal Inflammation In Infants Epigenetic Changes In Multiple Sclerosis – Studied In Twins
Epigenetic Changes In Multiple Sclerosis – Studied In Twins Did You Know Math Can Help Us Learn How Diseases Work?
Did You Know Math Can Help Us Learn How Diseases Work?