





Following a meal, the hormone insulin exerts well-known endocrine effects on the body by stimulating energy uptake in tissues to regulate metabolism. In Drosophila, this same signal is also closely monitored by cells of the so-called “stem cell niche,” a protective regulatory microenvironment surrounding stem cells. The niche controls proliferation and differentiation of stem cells through the deposition of matrix molecules and local secretion of specific growth factors. By listening to insulin signals, the niche can coordinate local tissue responses to changing environmental conditions by regulating stem behavior.
In our laboratory, we have recently identified similar insulin-dependent control of stem cells by their niche in the mammalian liver during injury. Interestingly, the short-range communication between stem cells and their niches broke down as a consequence of “insulin resistance,” a systemic state of reduced insulin sensitivity affecting patients with obesity and type II diabetes. Our study, therefore, predicts that the capacity for tissue repair is diminished in the liver by an endocrine disorder that is intimately associated with the toxic effects of a Western diet and lifestyle.
Our investigation set out to test the impact of insulin resistance on liver repair. One of our most regenerative organs, the liver is capable of sustaining injury caused by drugs, toxins, infections, or chronic disease. Often, in response to chronic damage, the liver undergoes a process of tissue remodeling, during which cells are reorganized and both scar tissue and new epithelial cells are generated (Figure 1).
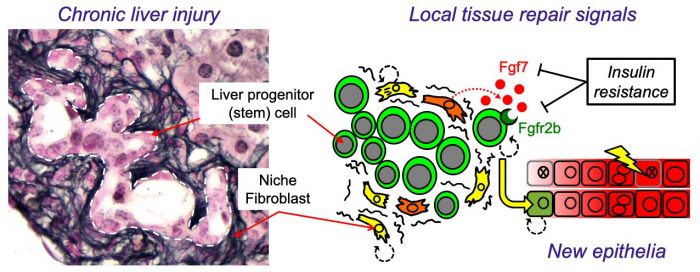
Figure 1 – Insulin resistance produced a less nurturing microenvironment for liver stem cells. (Left) Injured mouse liver showing expansion of bipotent liver progenitor (stem) cells (LPCs) within duct-like structures, surrounded by cells of the fibrotic niche (stained black: Reticulin). (Right) Cartoon illustrating how local control LPCs by niche fibroblasts is disrupted by insulin resistance resulting in reduced FGF7 expression, loss of the receptor (FGFR2b) in LPCs and a resulting defect in liver repair. Image courtesy Luke Noon.
Liver function is maintained by the production of new epithelial cells (hepatocytes and cholangiocytes) from a pool of stem cells called “liver progenitor cells” (LPCs). Interestingly LPC expansion, and hence the adaptive injury-response, is driven by factors produced by fibroblasts; a cell type too often considered the “bad guys” of liver disease because their excessive activation results in scar tissue (liver fibrosis). However, fibroblasts also produce important regenerative signals such as FGF7 (Fibroblast growth factor 7), known for its ability to stimulate epithelial repair in the skin during wound healing. Because of the peculiar biology of FGF7 signaling, this local growth factor exerts exquisitely directed short-range effects upon adjacent LPCs expressing its receptor (FGFR2b). Mice engineered without FGF7 have a severely impaired response to liver injury because LPCs are no longer triggered to react.
In our study, using insulin-resistant mice, we found that fibroblasts of the niche were unable to sustain FGF7 expression during chronic liver injury, whilst LPCs also failed to appropriately express the receptor (FGFR2b). As a consequence, niche-to-LPC FGF7 signaling was reduced, and livers were less able to undergo adaptive tissue remodeling during injury. A similar dual-loss of both signal and receptor was also observed in vitro using human cells in a tissue culture model of liver injury. These experiments confirmed that insulin signals are sensed by niche cells and that this important information is relayed to adjacent LPCs via FGF7, stimulating their differentiation and the production of new epithelia.
An interesting twist in our story was the observation that, in addition to being less attentive hosts towards their LPC neighbors, insulin-resistant niche cells were also more active in producing a fibrotic microenvironment, suggesting that normal insulin signals encourage a healthy balance between the necessary fibrotic response and epithelial repair. Our results, therefore, give new insight into the known association between insulin resistance and liver scarring, whilst also providing a mechanistic framework towards a greater understanding of the gradual progression of liver disease in patients with underlying metabolic dysfunction.
In more general terms, our study sets a novel precedent for how adult stem cell behavior in mammalian tissues can be regulated by insulin. The highly conserved insulin signal, whilst generic and addressed to all cells via circulation, provides important global information about energy status and feeding behavior. That such information should help inform the local decision-making process performed by stem cells and their niche during prolonged periods of injury seems logical. We speculate that under conditions of starvation when insulin levels are reduced, the tailoring of wound repair to favor short term scaring over the rebuilding of tissue could have evolutionary advantages in terms of survival. In contrast, insulin resistance, which starves the tissues of normal insulin signaling can be considered a maladaptive response to the excesses of Western diet – with the potential to limit the regenerative capacity of vital organs such as the liver. Insulin resistance is associated with accelerated aging and chronic disease in vital organs such as the heart, the vasculature, the liver, and the brain. However, it remains to be seen whether a “niche” role for insulin underlies pathogenic aspects of the metabolic syndrome in human patients.
These findings are described in the article entitled Insulin resistance disrupts epithelial repair and niche-progenitor Fgf signaling during chronic liver injury, recently published in the journal PLOS Biology.
Related Posts
 Important Strategies To Help Healthcare Providers Support Patients With Diabetes
Important Strategies To Help Healthcare Providers Support Patients With Diabetes Studying The Link Between Increased BMI And Late-Onset Preeclampsia In Pregnant Women
Studying The Link Between Increased BMI And Late-Onset Preeclampsia In Pregnant Women A Bacterial Cell Imaging Method Using CRISPR And Microfluidics
A Bacterial Cell Imaging Method Using CRISPR And Microfluidics B. infantis Reduces Key Markers Of Intestinal Inflammation In Infants
B. infantis Reduces Key Markers Of Intestinal Inflammation In Infants Epigenetic Changes In Multiple Sclerosis – Studied In Twins
Epigenetic Changes In Multiple Sclerosis – Studied In Twins Did You Know Math Can Help Us Learn How Diseases Work?
Did You Know Math Can Help Us Learn How Diseases Work?