





Pompe disease is a rare and potentially fatal metabolic myopathy (prevalence 1 in 20,000). In Pompe disease, the deficiency of lysosomal acid α-glucosidase (GAA) leads to accumulation of lysosomal glycogen and muscle damage. Enzyme replacement therapy (ERT) for Pompe disease (glycogen storage disease type II; acid maltase deficiency) with alglucosidase alfa (rhGAA; LumizymeR) was approved by the Food and Drug Administration in 2006.
Although treatment with ERT can slow the progression of symptoms in Pompe disease patients, the limitations of this ERT are significant, including high dosages and incomplete reversal of muscle weakness. New treatments are urgently needed for patients with Pompe disease, including for adults with late-onset Pompe disease (LOPD). Our experiments in mice with Pompe disease showed that increasing the uptake of rhGAA by the cation-independent mannose-6-phosphate receptor (CI-MPR)-mediated uptake of rhGAA by treatment with a β2 agonist drug increased the benefits from ERT. Our recently published Phase I/II clinical study with the β2 agonist clenbuterol showed improvements in the 6-minute walk test (6MWT), a validated endpoint in LOPD (Koeberl et al 2018).
Experiments showed benefits from clenbuterol treatment
Effective dosages for ERT in Pompe disease are up to 100-fold greater than those in other lysosomal disorders. This high-dose requirement has been attributed to the low amount of the receptor for rhGAA in skeletal muscle, where it is needed for the uptake of rhGAA during ERT (Fig. 1A).
Our experiments revealed benefits from increasing the uptake rhGAA during ERT in GAA knockout (KO) mice with Pompe disease (Koeberl et al 2012; Koeberl et al 2011). These studies revealed that clenbuterol, a selective β2 agonist, enhanced CI-MPR expression and increased efficacy from ERT, thereby confirming the key role of CI-MPR with regard to ERT in Pompe disease (Koeberl et al 2012; Koeberl et al 2011). The amount of stored glycogen in muscle was decreased by clenbuterol treatment during ERT (Fig. 1B).
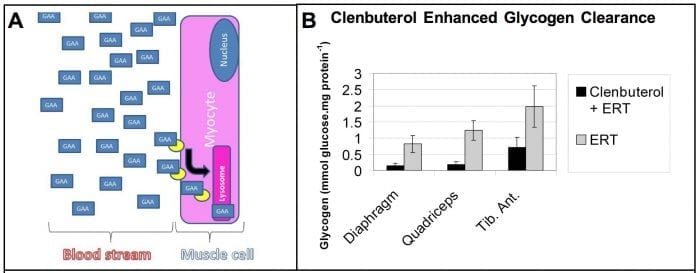
Figure 1. ERT depends upon receptor-mediated uptake of recombinant lysosomal enzymes. (A) In Pompe disease the CI-MPR (yellow oval) binds GAA (blue box) at the cell membrane and traffics it to the lysosome (pink box), and therefore a drug that increased CI-MPR would enhance biochemical correction from ERT. (B) A selective 2-agonist, clenbuterol, increased CI-MPR expression and significantly enhanced biochemical correction in combination with ERT, in comparison with ERT alone, as demonstrated by decreased glycogen storage in mice with a classical lysosomal storage disorder, Pompe disease (Koeberl et al 2011). Credit: Dwight Koeberl
Importantly, skeletal muscles comprised primarily of type II myofibers, including the tibialis anterior muscle, responded better to ERT following clenbuterol treatment, (Koeberl et al 2011). This is important because type II muscles are resistant to ERT due to low CI-MPR expression (Raben et al 2003; Raben et al 2005). The underlying mechanism for the effects of clenbuterol on muscle is increased expression of CI-MPR (Matsumoto et al 2006). Clenbuterol also caused muscle hypertrophy, which could be beneficial in Pompe disease (Matsumoto et al 2006).
Unmet clinical need in Pompe disease
The availability of ERT has prolonged the survival of patients with infantile-onset Pompe disease, which has increased the understanding of pathology and extent of disease in these patients (Kishnani et al 2007). Even in patients with a good response to ERT, however, residual muscle weakness has been observed (Jones et al 2010; Nicolino et al 2009; Yanovitch et al 2010). Importantly, the combination of clenbuterol treatment with ERT reversed neuromuscular involvement in GAA-KO mice with Pompe disease (Koeberl et al 2012; Koeberl et al 2011).
Previously, low levels of CI-MPR were demonstrated in skeletal muscle of GAA-KO mice, specifically in muscles comprised primarily of type II myofibers (Raben et al 2003; Raben et al 2005). The importance of CI-MPR expression to ERT in Pompe disease was demonstrated by the enhanced efficacy of modified rhGAA – engineered to increase the number of mannose-6-phosphate moieties to increase binding with CI-MPR (McVie-Wylie et al 2008; Zhu et al 2009). Consistent with our prediction that increased CI-MPR will enhance the uptake and lysosomal targeting of GAA, Pompe disease patient fibroblasts were found to be deficient in CI-MPR recycling and uptake of rhGAA was impaired (Cardone et al 2008).
Furthermore, our earlier open-label clinical trial with another β2 agonist drug, albuterol, combined with ERT in adults with LOPD revealed safety and improved muscle function (Koeberl et al 2014). However, albuterol has been less effective at lowering muscle glycogen than clenbuterol in our preclinical experiments (Koeberl et al 2012; Li et al 2013), and adjunctive albuterol did not decrease muscle glycogen in our pilot clinical trial (Koeberl et al 2014).
Clinical trial with clenbuterol in LOPD
We recently published a clinical trial of clenbuterol treatment in adult patients with LOPD (Koeberl et al 2018). The goal of this study was to investigate whether adjunctive clenbuterol therapy could improve the benefits of ERT in Pompe disease. In this 52-week clinical trial, we investigated the use of clenbuterol in patients with LOPD who were stably treated with ERT for >3 years. Only minor side effects were caused by clenbuterol treatment, including transient tremors and cramping, and mild increases in the muscle enzyme, creatine kinase.
Benefits at Week 52 included increased distance walked in the 6MWT, which measures how far someone can walk on a standard track in 6 minutes. The strength of breathing muscles also increased. Specialized muscle function testing also showed benefits in patients taking clenbuterol Muscle biopsies at the beginning and end of the study revealed lower glycogen in the muscle of patients taking clenbuterol, but not in the muscle of patients taking a placebo (a sugar pill). Sequencing of muscle RNA revealed beneficial changes in genes that are active in muscle only for patients taking clenbuterol. In summary, this study revealed initial safety and benefits from clenbuterol treatment in patients with LOPD who are stably treated with ERT, long after the benefits from ERT had stabilized or declined.
We propose to continue investigating the use of a selective β2 agonist in combination with ERT in order to progress to late-stage trials in LOPD. We predict that clenbuterol therapy will enhance the response to ERT in Pompe disease and other lysosomal storage disorders. Furthermore, increasing CI-MPR expression could reduce the dosage requirements for ERT or a future gene therapy (Li et al 2013). Overall, the availability of treatments that can prove the efficacy of ERT for Pompe disease and other lysosomal storage disorders will improve efficacy and reduce the costs of therapies for these diseases.
These findings are described in the article entitled Correction of Biochemical Abnormalities and Improved Muscle Function in a Phase I/II Clinical Trial of Clenbuterol in Pompe, recently published in the journal Molecular Therapy. This work was conducted by Dwight D. Koeberl, Laura E. Case, Edward C. Smith, Crista Walters, Sang-oh Han, Yanzhen Li, Wei Chen, Christoph P. Hornik, Kim M. Huffman, William E. Kraus, Beth L. Thurberg, David L. Corcoran, Deeksha Bali, Nenad Bursac, and Priya Kishnani from Duke University.
References:
- Cardone, M, Porto, C, Tarallo, A, et al. (2008). Abnormal mannose-6-phosphate receptor trafficking impairs recombinant alpha-glucosidase uptake in Pompe disease fibroblasts. Pathogenetics 1: 6.
- Jones, HN, Muller, CW, Lin, M, et al. (2010). Oropharyngeal dysphagia in infants and children with infantile Pompe disease. Dysphagia 25: 277-283.
- Kishnani, PS, Corzo, D, Nicolino, M, et al. (2007). Recombinant human acid [alpha]-glucosidase: major clinical benefits in infantile-onset Pompe disease. Neurology 68: 99-109.
- Koeberl, DD, Austin, S, Case, LE, et al. (2014). Adjunctive albuterol enhances the response to enzyme replacement therapy in late-onset Pompe disease. FASEB J 28: 2171-2176.
- Koeberl, DD, Case, LE, Smith, EC, et al. (2018). Correction of Biochemical Abnormalities and Improved Muscle Function in a Phase I/II Clinical Trial of Clenbuterol in Pompe Disease. Mol Ther 26: 2304-2314.
- Koeberl, DD, Li, S, Dai, J, Thurberg, BL, Bali, D, and Kishnani, PS. (2012). Beta2 Agonists enhance the efficacy of simultaneous enzyme replacement therapy in murine Pompe disease. Mol Genet Metab 105: 221-227.
- Koeberl, DD, Luo, X, Sun, B, et al. (2011). Enhanced efficacy of enzyme replacement therapy in Pompe disease through mannose-6-phosphate receptor expression in skeletal muscle. Mol Genet Metab 103: 107-112.
- Li, S, Sun, B, Nilsson, MI, et al. (2013). Adjunctive beta2-agonists reverse neuromuscular involvement in murine Pompe disease. FASEB J 27: 34-44.
- Matsumoto, T, Akutsu, S, Wakana, N, Morito, M, Shimada, A, and Yamane, A. (2006). The expressions of insulin-like growth factors, their receptors, and binding proteins are related to the mechanism regulating masseter muscle mass in the rat. Arch Oral Biol 51: 603-611.
- McVie-Wylie, AJ, Lee, KL, Qiu, H, et al. (2008). Biochemical and pharmacological characterization of different recombinant acid alpha-glucosidase preparations evaluated for the treatment of Pompe disease. MolGenetMetab 94: 448-455.
- Nicolino, M, Byrne, B, Wraith, JE, et al. (2009). Clinical outcomes after long-term treatment with alglucosidase alfa in infants and children with advanced Pompe disease. Genet Med 11: 210-219.
- Raben, N, Danon, M, Gilbert, AL, et al. (2003). Enzyme replacement therapy in the mouse model of Pompe disease. Molecular Genetics and Metabolism 80: 159-169.
- Raben, N, Fukuda, T, Gilbert, AL, et al. (2005). Replacing acid alpha-glucosidase in Pompe disease: Recombinant and transgenic enzymes are equipotent, but neither completely clears glycogen from type II muscle fibers. Molecular Therapy 11: 48-56.
- Yanovitch, TL, Banugaria, SG, Proia, AD, and Kishnani, PS. (2010). Clinical and histologic ocular findings in pompe disease. J Pediatr Ophthalmol Strabismus 47: 34-40.
- Zhu, Y, Jiang, JL, Gumlaw, NK, et al. (2009). Glycoengineered Acid alpha-Glucosidase With Improved Efficacy at Correcting the Metabolic Aberrations and Motor Function Deficits in a Mouse Model of Pompe Disease. Mol Ther 17: 954-63.
Related Posts
 Important Strategies To Help Healthcare Providers Support Patients With Diabetes
Important Strategies To Help Healthcare Providers Support Patients With Diabetes Studying The Link Between Increased BMI And Late-Onset Preeclampsia In Pregnant Women
Studying The Link Between Increased BMI And Late-Onset Preeclampsia In Pregnant Women A Bacterial Cell Imaging Method Using CRISPR And Microfluidics
A Bacterial Cell Imaging Method Using CRISPR And Microfluidics B. infantis Reduces Key Markers Of Intestinal Inflammation In Infants
B. infantis Reduces Key Markers Of Intestinal Inflammation In Infants Epigenetic Changes In Multiple Sclerosis – Studied In Twins
Epigenetic Changes In Multiple Sclerosis – Studied In Twins Did You Know Math Can Help Us Learn How Diseases Work?
Did You Know Math Can Help Us Learn How Diseases Work?