




Cardiovascular regeneration focuses on repairing or replacing damaged or senescent cardiac and vascular tissue. This damage is largely caused by myocardial ischemia (poor perfusion of the heart causing dysfunction or loss of cardiac tissue) and fibrosis (with replacement of myocytes and vessels with non-functional scar tissue), each of which may lead to heart failure, impaired functional capacity and quality of life, serious arrhythmias, and death.
Harnessing the patient’s own reparative capacity to produce new and healthy tissue is one of the goals of regenerative medicine. The field of regenerative medicine was galvanized in 2006 with the discovery of the major regulators of pluripotency. Six years later, Shinya Yamanaka won the Nobel Prize for his discovery of these pluripotency factors (OCT4, SOX2, KLF4 and cMYC) (1, 2). Pluripotency is the ability of a cell to generate any somatic cell lineage. We have shown that in addition to the Yamanaka factors, innate immune activation is required for efficient nuclear reprogramming to pluripotency (3). As described below, we have made progress in delineating the mechanisms by which innate immune signaling increases epigenetic plasticity and enhances nuclear reprogramming, a process we have termed “transflammation” (3).
Yamanaka’s work led to the concept that the forced expression of the master regulators of cell identity might be used to transdifferentiate a somatic cell to any other lineage. Indeed, his work led to an intensive search for factors whose forced expression could change one type of cell into another lineage. For example, a number of groups have reported that fibroblasts may be converted to other lineages, including neurons, cardiomyocytes and endothelial cells, using viral vectors encoding master regulators of cell identity (4-6). Our knowledge of the role of innate immune signaling in nuclear reprogramming has permitted us to develop a non-viral method to transdifferentiate human fibroblasts into endothelial cells, and to show their utility in a mouse model of limb ischemia (7).
Transflammation is the process by which innate immune signaling promotes epigenetic plasticity, which is a global change in the epigenetic machinery of a cell required for it to transform to another cell type (Figure 1). In our recent paper titled, “Transflammation: Innate Immune Signaling in Nuclear Reprogramming” (8)we reviewed the concepts of innate immune signaling, as it pertains to nuclear reprogramming to pluripotency, to transdifferentiation, and to tissue regeneration. As we continue to explore the mechanisms of transflammation and innate immunity, we are hopeful that we will forge a path toward developing new therapeutic avenues for regenerative medicine.
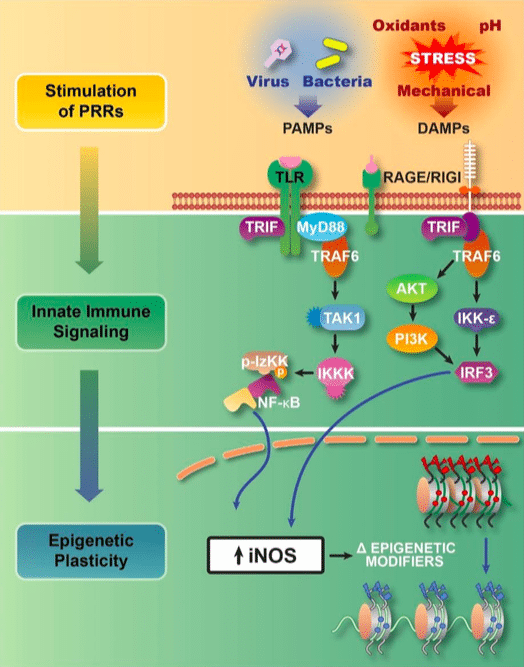
Figure 1:Transflammation is an adaptive response to cellular challenges. Pathogen associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) activate pattern recognition receptors (PRRs) such as the toll-like receptors (TLRs) and the receptor for advanced glycation end products (RAGE) which trigger innate immune signaling that leads to the activation of NFκB and IRF-3. These transcriptional factors cause global changes in the expression of epigenetic modifiers (upregulation of histone methyltransferases, and downregulation of Dot1L and histone deacetylases). In addition, the expression of inducible nitric oxide synthase leads to the S-nitrosylation of epigenetic modifiers to alter their activity and association with the chromatin. These changes in the expression and activity of epigenetic modifiers increase the open probability state of the chromatin. The increase in DNA accessibility provides for phenotypic fluidity, and adaptive responses to the cellular challenge (8). (Republished with permission from Elsevier)
Innate immunity is the intrinsic cellular response to foreign pathogens. Cellular recognition of these pathogens triggers a cell-autonomous response by initiating a signaling cascade that triggers the synthesis and release of cytokines. We discovered that this response also triggers global changes in epigenetic modifiers which facilitate the nuclear reprogramming required during the induction of pluripotency and in transdifferentiation(3). Further, we determined that this immune response involves the activation of toll-like receptors (TLR) or RIG-1-like receptors (RLR) that trigger signaling cascades, resulting in NFκB or IRF-3 mediated changes in epigenetic plasticity (9).
Reactive oxygen species (ROS) and reactive nitrogen species (RNS) also play a role in regulating DNA accessibility (10, 11). We also observed that there is a “Goldilocks’ zone” for innate immunity activation in reprogramming (11)(Figure 2). While a modest stimulation of innate immune signaling (using low doses of Poly I:C) increases iPSC generation, higher doses decrease iPSC generation without impairing cell viability.
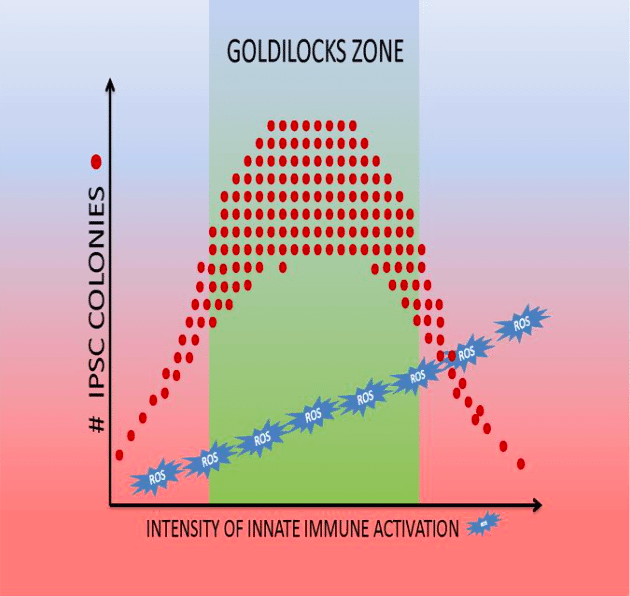
Figure 2:Goldilocks’ zone of innate immune signaling for the generation of iPSCs (9). (Republished with permission from Elsevier)
Transdifferentiation has typically been forced by using viral vectors to overexpress lineage determination factors (12, 13). We determined that because innate immune signaling increases epigenetic plasticity, it facilitates cellular reprogramming using external cues. Priming cells with Poly I:C, we found that the subsequent addition of endothelial growth factors could induce transdifferentiation of fibroblasts to endothelial cells (7). Thus, we were able to induce transdifferentiation without viral vectors encoding transcriptional factors. These fibroblast-derived endothelial cells (iECs) not only displayed all of the immunohistochemical markers of endothelial cells, but also possessed endothelial functions not present in fibroblasts (e.g. generation of nitric oxide, formation of tubular networks in matrigel; uptake of acetylated LDL) (9).
The transcriptional profile and histone modifications were very similar to authentic endothelial cells. Notably, in the absence of innate immune signaling precipitated by Poly I:C, endothelial growth factors alone could not transdifferentiate fibroblasts into endothelial cells. Furthermore, when we knocked down the expression of TLR3 in fibroblasts, these cells were resistant to our transdifferentiation protocol, as shown by lower yield of iECs, and the incomplete reprogramming of these iECs (less fidelity to the transcriptional, phenotypic and functional profile of endothelial cells). These studies reaffirmed the significant role of innate immunity in transdifferentiation (9).
Mammals have a limited ability to regenerate tissue in response to injury. While adult humans can partially regenerate the liver, skeletal muscle, and peripheral nervous system, the regenerative capacity of organs like the heart is limited (14). The process of tissue regeneration is complex and requires multiple cell types. The role of immune cells in regeneration has been well reviewed (15, 16), but the role of the cell-autonomous innate immune signaling is less studied. Our work suggests that cell-autonomous innate immune signaling is the first response to injury, causing global changes in the activity and expression of epigenetic modifiers that favor phenotypic fluidity in non-immune cells. Our observation of a Goldilocks’ zone also reconciles the conflicting literature reports on the role of innate immune signaling in organ regeneration and tissue repair (Table 1), suggesting an optimal level of activation is required for efficient reprogramming.
| Mice | Regenerative phenotype |
| Tlr3-/- | Delayed skin wound healing (17)
Decreased angiogenesis and arteriogenesis in hindlimb ischemia model (18) |
| Tlr4-/- | Reduced cardiac hypertrophy following pressure overload (19)
Delayed skin wound healing (20) |
| Tlr2-/- | Delayed intestine regeneration (21) |
| Tlr4-/- Tlr2-/- | Impaired lung repair after acute lung injury (22) |
| MyD88-/- | Delayed skin wound healing (14)
Impaired lung repair after acute lung injury (22) Delayed liver regeneration (23) Delayed intestine regeneration (21) |
| Ifnar1-/- MyD88-/- | Normal liver regeneration (24) |
| Infγ-/- | Promoted skin wound healing (25) |
| Il6-/- | Defective liver regeneration (26) |
Table 1. Regenerative phenotype of mice deficient of key innate immune signaling molecules (Republished with permission from Elsevier)
We have unpublished data that show both insufficient activation of innate immune signaling and excessive activation of innate immune signaling impair nuclear reprogramming. This observation has also been seen in clinic settings. Patients treated with steroids have reduced innate immune signaling, and have impaired wound healing. In contrast, diabetic patients with excessive and chronic inflammation of their foot ulcers also have reduced wound healing.
In conclusion, we have found that innate immune signaling causes global changes in the expression and activity of epigenetic modifiers required for nuclear reprogramming, transdifferentiation, and tissue regeneration. Optimal activation of innate immunity increases epigenetic plasticity that enhances phenotypic fluidity in a process we have termed transflammation. We believe that understanding the role of transflammation and innate immune mechanisms in tissue regeneration and repair will lead us into a new frontier of regenerative medicine and to eventual treatments that will enhance the body’s ability to heal itself.
Acknowledgment: This work was supported in part by a grant to JPC from the NHLBIR01 HL133254.
These findings are described in the article entitled Transflammation: Innate immune signaling in nuclear reprogramming, recently published in the journal Advanced Drug Delivery Reviews. This work was conducted by Shu Meng, Palas Chanda, Rajarajan A. Thandavarayan, and John P. Cooke from the Houston Methodist Research Institute.
References:
1. Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131(5):861-72. doi: 10.1016/j.cell.2007.11.019. PubMed PMID: 18035408.
2. Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126(4):663-76. doi: 10.1016/j.cell.2006.07.024. PubMed PMID: 16904174.
3. Lee J, Sayed N, Hunter A, Au KF, Wong WH, Mocarski ES, Pera RR, Yakubov E, Cooke JP. Activation of innate immunity is required for efficient nuclear reprogramming. Cell. 2012;151(3):547-58. doi: 10.1016/j.cell.2012.09.034. PubMed PMID: 23101625; PMCID: PMC3506423.
4. Qian L, Huang Y, Spencer CI, Foley A, Vedantham V, Liu L, Conway SJ, Fu JD, Srivastava D. In vivo reprogramming of murine cardiac fibroblasts into induced cardiomyocytes. Nature. 2012;485(7400):593-8. doi: 10.1038/nature11044. PubMed PMID: 22522929; PMCID: PMC3369107.
5. Wernig M, Meissner A, Foreman R, Brambrink T, Ku M, Hochedlinger K, Bernstein BE, Jaenisch R. In vitro reprogramming of fibroblasts into a pluripotent ES-cell-like state. Nature. 2007;448(7151):318-24. doi: 10.1038/nature05944. PubMed PMID: 17554336.
6. Wong WT, Cooke JP. Therapeutic transdifferentiation of human fibroblasts into endothelial cells using forced expression of lineage-specific transcription factors. J Tissue Eng. 2016;7:2041731416628329. doi: 10.1177/2041731416628329. PubMed PMID: 27081470; PMCID: PMC4820020.
7. Sayed N, Wong WT, Ospino F, Meng S, Lee J, Jha A, Dexheimer P, Aronow BJ, Cooke JP. Transdifferentiation of human fibroblasts to endothelial cells: role of innate immunity. Circulation. 2015;131(3):300-9. doi: 10.1161/CIRCULATIONAHA.113.007394. PubMed PMID: 25359165; PMCID: PMC4309381.
8. Meng S, Chanda P, Thandavarayan RA, Cooke JP. Transflammation: Innate immune signaling in nuclear reprogramming. Adv Drug Deliv Rev. 2017;120:133-41. doi: 10.1016/j.addr.2017.09.010. PubMed PMID: 28916494; PMCID: PMC5705345.
9. Sayed N, Ospino F, Himmati F, Lee J, Chanda P, Mocarski ES, Cooke JP. Retinoic Acid Inducible Gene 1 Protein (RIG1)-Like Receptor Pathway Is Required for Efficient Nuclear Reprogramming. Stem Cells. 2017;35(5):1197-207. doi: 10.1002/stem.2607. PubMed PMID: 28276156; PMCID: PMC5426114.
10. Meng S, Zhou G, Gu Q, Chanda PK, Ospino F, Cooke JP. Transdifferentiation Requires iNOS Activation: Role of RING1A S-Nitrosylation. Circ Res. 2016;119(9):e129-e38. doi: 10.1161/CIRCRESAHA.116.308263. PubMed PMID: 27623813; PMCID: PMC5065398.
11. Zhou G, Meng S, Li Y, Ghebre YT, Cooke JP. Optimal ROS Signaling Is Critical for Nuclear Reprogramming. Cell Rep. 2016;15(5):919-25. doi: 10.1016/j.celrep.2016.03.084. PubMed PMID: 27117405; PMCID: PMC4856580.
12. Huang P, He Z, Ji S, Sun H, Xiang D, Liu C, Hu Y, Wang X, Hui L. Induction of functional hepatocyte-like cells from mouse fibroblasts by defined factors. Nature. 2011;475(7356):386-9. doi: 10.1038/nature10116. PubMed PMID: 21562492.
13. Wada R, Muraoka N, Inagawa K, Yamakawa H, Miyamoto K, Sadahiro T, Umei T, Kaneda R, Suzuki T, Kamiya K, Tohyama S, Yuasa S, Kokaji K, Aeba R, Yozu R, Yamagishi H, Kitamura T, Fukuda K, Ieda M. Induction of human cardiomyocyte-like cells from fibroblasts by defined factors. Proc Natl Acad Sci U S A. 2013;110(31):12667-72. doi: 10.1073/pnas.1304053110. PubMed PMID: 23861494; PMCID: PMC3732928.
14. Stoick-Cooper CL, Moon RT, Weidinger G. Advances in signaling in vertebrate regeneration as a prelude to regenerative medicine. Genes Dev. 2007;21(11):1292-315. doi: 10.1101/gad.1540507. PubMed PMID: 17545465.
15. Forbes SJ, Rosenthal N. Preparing the ground for tissue regeneration: from mechanism to therapy. Nat Med. 2014;20(8):857-69. doi: 10.1038/nm.3653. PubMed PMID: 25100531.
16. Poss KD. Advances in understanding tissue regenerative capacity and mechanisms in animals. Nat Rev Genet. 2010;11(10):710-22. doi: 10.1038/nrg2879. PubMed PMID: 20838411; PMCID: PMC3069856.
17. Lin Q, Fang D, Fang J, Ren X, Yang X, Wen F, Su SB. Impaired wound healing with defective expression of chemokines and recruitment of myeloid cells in TLR3-deficient mice. Journal of immunology. 2011;186(6):3710-7. doi: 10.4049/jimmunol.1003007. PubMed PMID: 21317384.
18. Holfeld J, Tepekoylu C, Reissig C, Lobenwein D, Scheller B, Kirchmair E, Kozaryn R, Albrecht-Schgoer K, Krapf C, Zins K, Urbschat A, Zacharowski K, Grimm M, Kirchmair R, Paulus P. Toll-like receptor 3 signalling mediates angiogenic response upon shock wave treatment of ischaemic muscle. Cardiovascular research. 2016;109(2):331-43. doi: 10.1093/cvr/cvv272. PubMed PMID: 26676850.
19. Ha T, Li Y, Hua F, Ma J, Gao X, Kelley J, Zhao A, Haddad GE, Williams DL, William Browder I, Kao RL, Li C. Reduced cardiac hypertrophy in toll-like receptor 4-deficient mice following pressure overload. Cardiovascular research. 2005;68(2):224-34. doi: 10.1016/j.cardiores.2005.05.025. PubMed PMID: 15967420.
20. Chen L, Guo S, Ranzer MJ, DiPietro LA. Toll-like receptor 4 has an essential role in early skin wound healing. The Journal of investigative dermatology. 2013;133(1):258-67. doi: 10.1038/jid.2012.267. PubMed PMID: 22951730; PMCID: 3519973.
21. Scheeren FA, Kuo AH, van Weele LJ, Cai S, Glykofridis I, Sikandar SS, Zabala M, Qian D, Lam JS, Johnston D, Volkmer JP, Sahoo D, van de Rijn M, Dirbas FM, Somlo G, Kalisky T, Rothenberg ME, Quake SR, Clarke MF. A cell-intrinsic role for TLR2-MYD88 in intestinal and breast epithelia and oncogenesis. Nature cell biology. 2014;16(12):1238-48. doi: 10.1038/ncb3058. PubMed PMID: 25362351.
22. Jiang D, Liang J, Fan J, Yu S, Chen S, Luo Y, Prestwich GD, Mascarenhas MM, Garg HG, Quinn DA, Homer RJ, Goldstein DR, Bucala R, Lee PJ, Medzhitov R, Noble PW. Regulation of lung injury and repair by Toll-like receptors and hyaluronan. Nature medicine. 2005;11(11):1173-9. doi: 10.1038/nm1315. PubMed PMID: 16244651.
23. Seki E, Tsutsui H, Iimuro Y, Naka T, Son G, Akira S, Kishimoto T, Nakanishi K, Fujimoto J. Contribution of Toll-like receptor/myeloid differentiation factor 88 signaling to murine liver regeneration. Hepatology. 2005;41(3):443-50. doi: 10.1002/hep.20603. PubMed PMID: 15723296.
24. Vaquero J, Riehle KJ, Fausto N, Campbell JS. Liver Regeneration after Partial Hepatectomy Is Not Impaired in Mice with Double Deficiency of Myd88 and IFNAR Genes. Gastroenterology research and practice. 2011;2011:727403. doi: 10.1155/2011/727403. PubMed PMID: 22216024; PMCID: 3246728.
25. Ishida Y, Kondo T, Takayasu T, Iwakura Y, Mukaida N. The essential involvement of cross-talk between IFN-gamma and TGF-beta in the skin wound-healing process. Journal of immunology. 2004;172(3):1848-55. PubMed PMID: 14734769.
26. Cressman DE, Greenbaum LE, DeAngelis RA, Ciliberto G, Furth EE, Poli V, Taub R. Liver failure and defective hepatocyte regeneration in interleukin-6-deficient mice. Science. 1996;274(5291):1379-83. PubMed PMID: 8910279.
Related Posts
 Important Strategies To Help Healthcare Providers Support Patients With Diabetes
Important Strategies To Help Healthcare Providers Support Patients With Diabetes Studying The Link Between Increased BMI And Late-Onset Preeclampsia In Pregnant Women
Studying The Link Between Increased BMI And Late-Onset Preeclampsia In Pregnant Women A Bacterial Cell Imaging Method Using CRISPR And Microfluidics
A Bacterial Cell Imaging Method Using CRISPR And Microfluidics B. infantis Reduces Key Markers Of Intestinal Inflammation In Infants
B. infantis Reduces Key Markers Of Intestinal Inflammation In Infants Epigenetic Changes In Multiple Sclerosis – Studied In Twins
Epigenetic Changes In Multiple Sclerosis – Studied In Twins Did You Know Math Can Help Us Learn How Diseases Work?
Did You Know Math Can Help Us Learn How Diseases Work?