





Published by Kyle O’Connell
National Museum of Natural History and the University of Texas at Arlington
These findings are described in the article entitled The effect of missing data on coalescent species delimitation and a taxonomic revision of whipsnakes (Colubridae: Masticophis), recently published in the journal Molecular Phylogenetics and Evolution (Molecular Phylogenetics and Evolution 127 (2018) 356-366). This work was led by Kyle O’Connell from the National Museum of Natural History and the University of Texas at Arlington, and by Eric Smith from the University of Texas at Arlington.
Knowing the number of species within a species group is essential for conservation because we have to know the number of species in a group to conserve them. It is also important for studying evolutionary process because our studies of how species form and change are improved if we have a more accurate understanding of the number of species.
Species delimitation is the use of multiple types of methods to provide support for hypotheses about species boundaries. The species delimitation processes encompasses two steps: the identification of potential species, and the testing of species boundaries. In this paper, we conduct these two steps using a group of North American snakes called coachwhips and whipsnakes (Genus Masticophis). To identify and test species boundaries, we gathered several types of data including external measurements of snake specimens, mitochondrial sequence data, and thousands of nuclear sequence markers.
Our choice of the three datasets was a balance of using old and new techniques. First, we included external measurements because these are how scientists identified species boundaries in these groups before the DNA era. In addition, external measurements are still used by many researchers, especially in cases where DNA is not available, as with old specimens. We also collected two kinds of DNA sequence data, the first being mitochondrial DNA, which is passed down only by the mother and also evolves much faster than nuclear DNA. Thus, we can use this mitochondrial DNA to identify recent evolutionary changes, and also differences in evolutionary history between males and females.
We also gathered thousands of nuclear DNA markers using a technique called double-digest restriction site associate DNA sequencing (ddRAD). The method works by randomly shearing the genome with enzymes and then sequencing all of the DNA fragments. By aligning the shared fragments across individuals, we could look for shared DNA changes within and between species. However, one challenge with this method is that when you analyze species that are distantly related, the DNA is different enough that it can shear in different places, thus causing you to end up with fewer DNA sequences at the end and a lot of missing data. We wanted to test how this missing sequence data affected our final estimates of species boundaries.
We focused our analyses on three species groups, the coachwhip (Masticophis flagellum), the neotropical whipsnake (Masticophis mentovarius), and the Sonoran whipsnake (Masticophis bilineatus; Figure 1). Coachwhips are distributed across the United States, and previously had six subspecies described. The neotropical whipsnake also has a wide range, ranging from northern Mexico all the way to Venezuela. This species has five recognized subspecies, one of which (Masticophis mentovarius striolatus) has been classified in the past based on external features as both a coachwhip and a neotropical whipsnake.
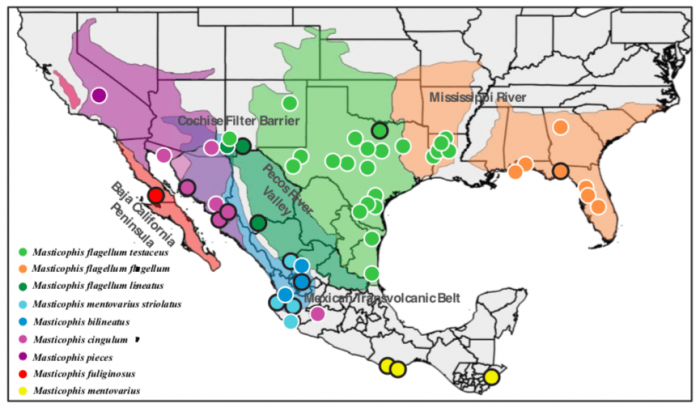
Figure 1. Map of whipsnake and coachwhip samples used in this study. Subspecies or species are labeled on the left, and the corresponding dots on the map represent individual DNA samples. Colors on the map show the previously described geographic range of the species or subspecies. Text on the map identifies geographical features that divide species. Credit: Kyle O’Connell
Finally, the Sonoran whipsnake only has two recognized subspecies and has a much more restricted range than coachwhips or neotropical whipsnakes. Very little recent work had been done on these snakes, and most of the species boundaries and relationships had been based on external scale counts and color patterns, but not recent DNA sequencing. One challenge with using only external features is that they may reflect adaptation to local environmental conditions rather than evolutionary history. DNA sequence should reflect the evolutionary history.
Exploring Species boundaries in coachwhips and whipsnakes
To identify species boundaries, we first looked at the external measurement data, much of which we had gathered from past studies. We found that external features would be used to identify species, particularly the number of belly scales and the color of the top of the animal. However, these features did not correspond to the evolutionary history, suggesting that local adaptation, rather than evolutionary history, may have caused these external differences.
Next, we estimated phylogenetic trees using the mitochondrial data. Phylogenetic trees are graphical representations of the evolutionary history based on DNA sequence similarity. We found that half of the recognized subspecies of coachwhips grouped more closely to the rest of the neotropical whipsnakes, and that the subspecies of neotropical whipsnake with a confusing past, Masticophis mentovarius striolatus, grouped more closely with the Sonoran whipsnake instead of the other neotropical whipsnakes! In other words, the historical understanding of species boundaries and relationships was not at all supported by the mitochondrial sequence data.
Finally, we conducted several types of analyses using the nuclear DNA data to further explore these species boundaries and relationships. In the nuclear data, however, we found that while there were probably several species of coachwhip, they were all related to one another and not related to the neotropical whipsnakes as in the mitochondrial data. However, Masticophis mentovarius striolatus still clustered closely with the Sonoran whipsnake. This suggested that each of those wide-ranging species may actually contain several undescribed species.
Testing hypotheses of species boundaries
Although thus far we had a good idea of the species boundaries and the evolutionary relationships between species, we still did not have any mathematical support for species boundaries. In other words, we wanted to place a number on how likely those species were to be “real.” In order to estimate this likelihood value, we used model-based tests, called coalescent methods, to provide support values for our species hypotheses.
For each species hypothesis, we ran the coalescent analysis, and at the end, it gave us an estimation for how likely that hypothesis is, based on the DNA sequence data we provided. For example, we compared the likelihood that eastern and western coachwhips were one species, or two, and at the end, had a numeric value to tell us which hypothesis was better supported. Now, this is the part where the missing data comes in because the likelihood calculation is only as good as the sequence data you provide. If you have five hundred genes, you have more information to use in the calculations. However, including more genes with ddRADseq data means more missing data, and it was unclear how these missing data would affect the likelihood calculations.
Finally, we ran the models for all combinations of species relationships with a lot of missing data (<50%) and less missing data (<20%). We found that the model supported dividing all of the subspecies we tested into independent species and that more missing data, which meant more genes included in the calculations, provided the same answers as less missing data. But including more genes provided higher support for the best model. In other words, all the analyses supported lots more species of whipsnakes and coachwhips than previously described, but the likelihood value for having more species was higher with more missing data because of the extra information included in the additional gene sequences. At the end of our paper, we chose to take a conservative approach and split coachwhips into an eastern and western species, divided at the transition between the Sonoran and Chihuahuan Deserts, and to elevate Masticophis mentovarius striolatus to Masticophis lineatus.
This study makes two primary contributions to science. First, it clarifies the species boundaries and relationships of coachwhips and whipsnakes. Conservation agencies may choose to manage these species differently now that species boundaries have been updated. Additionally, researchers studying evolutionary processes in this group will have a better idea of how they evolved by knowing where the species boundaries are. Second, our study found that having more missing data, particularly when using ddRADseq, allows you to include more DNA markers and thus increases the support for the best model. In other words, having a lot of missing data (or analysis noise) tends to be offset by the inclusion of more makers.
Related Posts
 Duck-Billed Dinosaurs Uncovered In Aniakchak, Alaska
Duck-Billed Dinosaurs Uncovered In Aniakchak, Alaska Cryptic Diversity In Vietnam’s Limestone Karst Habitats
Cryptic Diversity In Vietnam’s Limestone Karst Habitats An Improved Method To Remove Debris From Cyst Nematode Egg Suspensions And Computer-Aided Technologies For Egg Counting
An Improved Method To Remove Debris From Cyst Nematode Egg Suspensions And Computer-Aided Technologies For Egg Counting The Footprints Of Urbanization, Industrialization, And Agriculture On River Beds: Heavy Metal Contamination Assessment And Source Identification In River Sediments In Eastern China
The Footprints Of Urbanization, Industrialization, And Agriculture On River Beds: Heavy Metal Contamination Assessment And Source Identification In River Sediments In Eastern China Aging Dolphins Via Pectoral Flipper Radiography
Aging Dolphins Via Pectoral Flipper Radiography Glycoalkaloids In Potatoes: The Effect Of Biostimulants And Herbicides
Glycoalkaloids In Potatoes: The Effect Of Biostimulants And Herbicides