





Many cellular functions are mediated through interactions between protein molecules. Understanding specifics of these interactions, such as which part of a protein takes part in interactions and what proteins can interact with one other, can shed light on the molecular details of regulations and ultimately will provide a way to rationally modulate these interactions for biomedical intervention.
Identification of these binding interfaces has many important applications; it enables interfering with the protein activity, either to enhance or disrupt binding by designing suitable molecules. For instance, knowledge of the binding interface of proteins in the immune system has led to the development of several modern drugs including Nulojix, a recent immunosuppressive drug approved by the FDA to prevent transplant rejection.
The first step in this discovery process is to understand which part(s) of a protein can interact, i.e. to identify its binding interface. Proteins bind to small organic molecules as well as to other large protein molecules. While there has been much success in predicting the interaction sites between proteins and small molecules, the prediction accuracy of binding interfaces between two proteins is rather low. More than 150,000 protein structures have been solved experimentally to date, but only a small fraction of these proteins have a known complex structure, where binding interfaces can be directly identified. Even if a complex structure is known for a given protein it is likely to be only partially informative as proteins are assumed to have on average 3-10 interacting partners. It is, therefore, essential to develop computational methods that can bridge the knowledge gap and that can accurately predict the binding interfaces of proteins.
Computational methods developed thus far to predict binding interfaces can be classified under two broad categories – (1) methods that make use of prior knowledge of known interfaces between proteins referred to as homology-based1,2; and (2) ab initio methods3,4 that are based on identifying various interface specific characteristics of residue conservation, solvent accessibility, planarity, etc. Ab initio methods have the advantage that these can be used for any proteins without restriction, while homology-based approaches are more accurate but are limited to a subset of proteins for which a sequentially similar structure is known that has been experimentally solved in a complex form.
We introduced a docking-based, ab initio method5 that successfully predicts the binding interfaces of proteins that does not require prior knowledge of the protein’s binding partner. This result is similar to what has been previously observed for the binding of small molecules to proteins. In the case of small molecule binding, it was observed that there are certain “sticky sites” on the protein to which many small molecules bind, regardless of their nature.6,7 We have observed similar “Super Sites” on protein molecules that can be identified by docking a target to many different, biologically unrelated protein partners.
The binding sites are identified on a protein by docking the protein to many partner proteins of different sizes. We generate multiple docked poses for each partner. We then identify the amino acid residues on the protein that are within a short distance (usually 4 angstroms or less) of an atom in the partner protein. The frequency with which the amino acid residues are sampled at the interface in these docking experiments is analyzed. The most frequently sampled residues are considered to be the interface residues. Sufficient accuracy is achieved with about 200 docked poses and with three or more protein partners to account for the variability of possible interacting partners. Our method is of comparable accuracy to other methods that currently exist to identify the binding interface. The major advantage of this method is that the binding sites are identified without prior knowledge of the cognate partner.
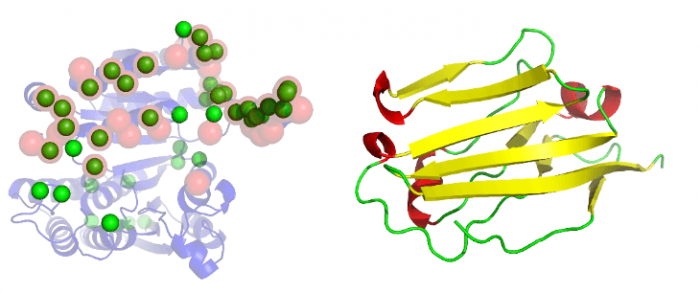
Figure 1. (A) Protein (1cnz.A) shown as a cartoon; annotated interface residues are shown as red transparent spheres; predicted residues shown as green spheres. (B) Partner protein (2jjs.C) used in the docking. Image republished with permission from PLOS Computational Biology from https://doi.org/10.1371/journal.pcbi.1006704
Our analysis revealed that the accuracy of the method is independent of the type of protein fold we are exploring and it appears to be a universal feature of protein. This is important because some protein folds are widely found in many biological processes, therefore, it is fair to assume that despite variability of functions, they preserved the mechanics of interactions to the same site. Our results show that docking to a generic partner is able to identify the likely binding site on a protein, which suggests that proteins have binding supersites, a favored site for interaction for both cognate and non-cognate ligands.
These findings are described in the article entitled Protein—protein binding supersites, recently published in the journal PLOS Computational Biology.
References:
- Petrey D, Honig B. Structural bioinformatics of the interactome. Annual review of biophysics, 2014;43:193-210.
- Zhang QC, Petrey D, Deng L, Qiang L, Shi Y, Thu CA, et al. Structure-based prediction of protein-protein interactions on a genome-wide scale. Nature, 2012; 490 (7421):556-60.
- Northey T, Baresic A, Martin ACR. IntPred: a structure-based predictor of protein-protein interaction sites. Bioinformatics, 2017.
- Porollo A, Meller J. Prediction-based fingerprints of protein-protein interactions. Proteins. 2007;66(3):630-645.
- Viswanathan R, Fajardo E, Steinberg G, Haller M, Fiser A. Protein binding supersites, PLoS Comput Biol 15(1): e1006704.
- Ringe D. what makes a binding site a binding site? Curr Opin Struct Biol. 1995;5(6):825-9.
- Hajduk PJ, Huth JR, Fesik SW. Druggability indices for protein targets derived from NMR-based screening data. J.Med.Chem. 2005;48(7):2518-25.
Related Posts
 Important Strategies To Help Healthcare Providers Support Patients With Diabetes
Important Strategies To Help Healthcare Providers Support Patients With Diabetes Studying The Link Between Increased BMI And Late-Onset Preeclampsia In Pregnant Women
Studying The Link Between Increased BMI And Late-Onset Preeclampsia In Pregnant Women A Bacterial Cell Imaging Method Using CRISPR And Microfluidics
A Bacterial Cell Imaging Method Using CRISPR And Microfluidics B. infantis Reduces Key Markers Of Intestinal Inflammation In Infants
B. infantis Reduces Key Markers Of Intestinal Inflammation In Infants Epigenetic Changes In Multiple Sclerosis – Studied In Twins
Epigenetic Changes In Multiple Sclerosis – Studied In Twins Did You Know Math Can Help Us Learn How Diseases Work?
Did You Know Math Can Help Us Learn How Diseases Work?