




Neurological disorders remain one of the last frontiers of medicine. Available treatments are few; of those that exist, most only mitigate symptoms and do nothing to slow the progression of the disease. Both the inaccessibility and the complexity of the brain pose major challenges to those who wish to develop disease-modifying therapies.
Take the molecular pathogenesis of even relatively straightforward Mendelian diseases such as Huntington’s disease (HD). The genetic cause is well understood: expansion of a CAG triplet repeat tract in the Huntingtin gene produces an abnormally long polyglutamine tract in the encoded protein.
Yet this single mutation affects thousands of other genes across numerous brain regions, in both neurons and glia. Transcriptomic, proteomic, and metabolomics studies yield an abundance of data on these changes, but some changes may reflect natural variation, environmental influences or even compensatory mechanisms. How are we to distinguish the alterations that drive disease pathogenesis from those that do not? If we could determine this, we might be able to devise an effective therapeutic approach that would subdue disease-driving pathways without dampening helpful compensatory responses.
Because Huntington’s disease is a single-gene disorder, it seemed to provide a good test case for developing a genetic algorithm that could then be applied to more complex cases such as the more common (and usually idiopathic) Alzheimer’s or Parkinson’s disease. Our main idea was to take validated gene expression alterations observed in human patients, then systematically mimic and/or antagonize those changes in a tractable model system to observe their effects.
We reasoned that recreating an alteration that drives the disease should aggravate neuronal dysfunction in the model, whereas recreating an alteration that reflects an attempt at compensating for the disease should mitigate the dysfunctions seen in the disease model. (We would expect that mimicking any other alteration that is not causal would have no effect on disease progression.)
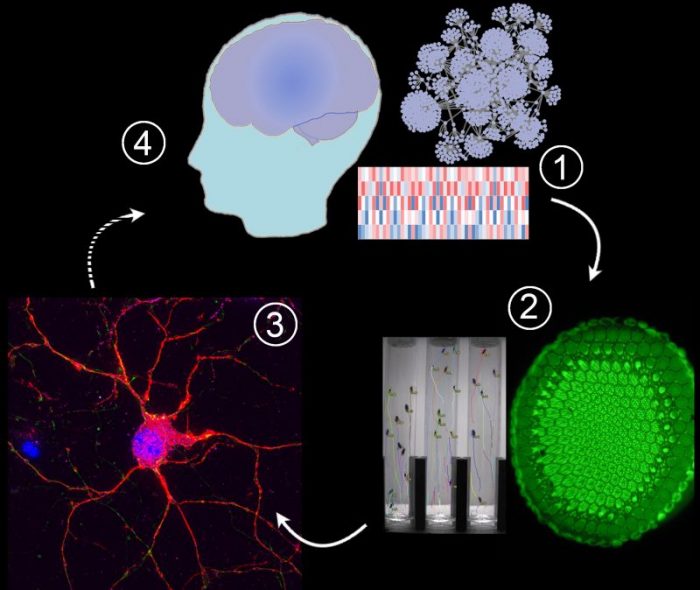
1) -Omic technologies reveal abnormal levels of thousands of mRNAs and proteins, but which of these alterations drive disease progression?
2) Disease-driving and compensatory alterations can be identified by integrating computational methods with gene perturbation analysis in Drosophila (fruit flies), a model system that allows high-throughput quantitative assessment of neuronal dysfunction. We replicate and/or antagonize each alteration in Drosophila brains to distinguish disease-drivers, compensatory, and non-causal alterations by observing how they affect the fruit fly model. In the Huntington’s disease studies we used the motor dysfunction assay because it is exquisitely sensitive and quantitative, but in some of our fruit fly models we can also express a mutant gene in the eye and assess the resulting dysmorphology in the ommatidia (large green disk in the photograph).
3) Disease drivers and compensatory alterations identified in fruit fly brains are validated in human neurons derived from pluripotent stem cells.
4) Disease driving and compensatory mechanisms can inform therapeutic interventions under development. Credit: Juan Botas
The choice of model system was a crucial one. The most physiologically relevant tests of gene expression alterations would be carried out in the context of an actual brain in a whole organism for which there are good, quantifiable readouts of neurological dysfunction. This leaves few options. We chose the fruit fly Drosophila because it’s genetically tractable — there were already knock-down and knock-up genetic tools to mimic gene expression alterations for the vast majority of the genes affected in HD — and experiments can be carried out on a large scale but within a short time-frame because we have a high-throughput, quantifiable behavioral assay — the climbing assay — that readily reveals neurodegeneration.
We narrowed down the list of genes to test by comparing transcriptomic data from both human Huntington’s disease patients and several different mouse models of the disease. (We reasoned that the more frequently a gene was reported to be altered across different species and studies, the more likely it was to be genuinely involved in HD.) We replicated and/or antagonized the transcriptional alterations for 312 genes in the neurons of HD model flies, who develop obvious impairment in the climbing assay by about two weeks of age. (Since mutant Huntingtin is expressed only in the neurons in these flies, their motor impairment is due to neurodegeneration and not a problem with their muscles.) Fruit flies show very robust negative geotaxis and when you tap them down inside a tube, they will quickly climb upward again. HD flies progressively lose their ability to climb as they suffer neurodegeneration, and their speed and paths can be analyzed using automated instruments.
This initial set of experiments yielded 44 transcriptomic alterations that further aggravated the neurodegeneration in HD flies and 38 that behaved as compensatory because they slowed down disease progression. Curious about possible functional connectivity among these 82 modifier genes, we constructed protein-protein interaction networks using the InWeb and String databases. This exercise revealed a subnetwork of genes implicated in cytoskeleton dynamics and inflammation whose dysregulation drives disease, as well as a second subnetwork of genes involved in Ca2+ homeostasis and vesicle biology whose dysregulation slows down disease progression. The 82 modifier genes have higher functional connectivity than would be expected at random (using as background either the whole genome or just genes normally expressed in a healthy striatum). We were able to validate these subnetworks by pathway extension, meaning that we found that other components of these pathways that were not among our original 312 genes were also disease modifiers.
Once the pathogenic/compensatory alterations and the corresponding pathways were firmly established in Drosophila, we turned to HD patient cells to further validate our findings. First, we used neurons derived from patient-derived stem cells to test a subset of the genes involved in inflammation and cytoskeleton dynamics. For these experiments, the readout was caspase-3 activation following BDNF deprivation (HD neurons are much more sensitive to BDNF deprivation and activate caspase, while healthy neurons do not). We then tested all 82 modifier genes in HD patient cells for their ability to modulate mutant Huntingtin (mHTT) protein levels, whose accumulation is the central pathogenic event in HD. (The mHTT protein is not cleared as readily from the cell as the wild-type protein, and the slow accumulation of the protein over time eventually overwhelms the cell’s normal functions.) We were curious to know if any of the modifier genes exerted their effects by modulating the stability or turnover of mHTT. Such modifiers would be particularly valuable, since lowering the levels of mHTT should be the most effective way to mitigate its many pathogenic downstream effects.
The data from HD patient cells revealed that a subset of modifier genes identified in Drosophila lower mHTT protein levels in HD patient cells, and at least some of them do so through the autophagic degradation system. This not only provides insight into the molecular genetics of disease progression in HD but points the way to specific targets that could be studied for therapeutic development. More importantly, the general approach can be used to distinguish pathogenic from compensatory “-omics” changes in other systems and other diseases.
These findings are described in the article entitled High-Throughput Functional Analysis Distinguishes Pathogenic, Nonpathogenic, and Compensatory Transcriptional Changes in Neurodegeneration, recently published in the journal Cell Systems.
This work was conducted by Ismael Al-Ramahi, Maria de Haro, Tatiana Gallego-Flores, Nazish T. Malik, Kelly Erikson, Benjamin A. Bleiberg, Matthew Avalos, George Fan, Laura Elizabeth Rivers, Javier R. Diaz-García, and Juan Botas from Baylor College of Medicine and Jan and Dan Duncan Neurological Research Institute at Texas Children’s Hospital, Boxun Lu from Fudan University, Simone Di Paola, Ivana Peluso, and Diego L. Medina from the Telethon Institute of Genetics and Medicine (TIGEM), Kaifang Pang, Andrew M. Laitman, and Zhandong Liu from the Jan and Dan Duncan Neurological Research Institute at Texas Children’s Hospital, and Marc Hild and James Palacino from the Novartis Institutes for Biomedical Research.
Related Posts
 Important Strategies To Help Healthcare Providers Support Patients With Diabetes
Important Strategies To Help Healthcare Providers Support Patients With Diabetes Studying The Link Between Increased BMI And Late-Onset Preeclampsia In Pregnant Women
Studying The Link Between Increased BMI And Late-Onset Preeclampsia In Pregnant Women A Bacterial Cell Imaging Method Using CRISPR And Microfluidics
A Bacterial Cell Imaging Method Using CRISPR And Microfluidics B. infantis Reduces Key Markers Of Intestinal Inflammation In Infants
B. infantis Reduces Key Markers Of Intestinal Inflammation In Infants Epigenetic Changes In Multiple Sclerosis – Studied In Twins
Epigenetic Changes In Multiple Sclerosis – Studied In Twins Did You Know Math Can Help Us Learn How Diseases Work?
Did You Know Math Can Help Us Learn How Diseases Work?