





Frontotemporal dementia (FTD) and amyotrophic lateral sclerosis (ALS) are two devastating neurodegenerative disorders. FTD groups different types of dementias affecting the frontal or temporal lobes of the brain. Depending on which region is most affected, FTD patients may suffer from speech and behavioral abnormalities, as well as dementia in later phases of the disease.
ALS, also known as Lou Gehrig’s disease, causes the death of motor neurons controlling voluntary muscle movement. This results in movement disorders and gradual weakening of the muscles, including those controlling eating, speaking and even breathing. Most ALS patients die within 3-5 years of diagnosis, although some may live longer, such as the famous physicist Stephen Hawking.
Although for most patients the cause of ALS/FTD is unknown, recent research discovered that a mutation in the C9ORF72 gene underlies a large fraction of the familial cases of both syndromes (DeJesus-Hernandez et al., 2011; Renton et al., 2011). The DNA code is written using four letters or “bases”: cytosine (C), guanine (G), adenine (A) or thymine (T). The C9ORF72 gene contains a GGGGCC sequence that is repeated a few times in healthy individuals, but up to several thousand times in C9ORF72 ALS/FTD patients.
One of the consequences of such massive expansion is the aberrant translation of the GGGGCC sequence into so-called dipeptide repeat proteins: poly-GlycineAlanine (poly-GA), poly-GlycineProline, poly-ProlineAlanine, poly-GlycineArginine, and poly-ProlineArginine. All of them form aggregates in the brain of C9ORF72 ALS/FTD patients, but most of these aggregates contain poly-GA (Edbauer and Haass, 2016). However, whether and how these aggregates contribute to neuronal death is poorly understood.
In a recent study, researchers from the Max Planck Institute of Biochemistry and the German Center for Neurodegenerative Diseases have investigated the pathological roles of poly-GA aggregates using cutting-edge electron microscopy (EM) techniques (Guo et al., 2018). EM allows visualizing the cell interior with much higher magnification than optical microscopes. But electrons can only propagate successfully in high vacuum, and therefore the samples must be inserted in the vacuum system for EM examination. This is complicated in the case of biological samples such as cells or tissues, which consist mainly of water.
Thus, to allow EM studies samples are traditionally prepared using a series of harsh procedures, including dehydration and heavy-metal staining. But groundbreaking work in the 1980s and 90s showed that biological samples in a close-to-native state can also be imaged by EM, provided that they are frozen fast enough that their water molecules do not have time to reorganize into ice crystals and remain in a glass-like vitreous state. This revolutionary technique, known as cryo-electron microscopy (cryo-EM), was awarded the Nobel Prize in Chemistry in 2017.
Thanks to further technological developments, it is now possible to 3D image very thin sections of cells, such as neurons, using a variant of cryo-EM known as cryo-electron tomography (cryo-ET) (Wagner et al., 2017). In this approach, cells are first quickly frozen to achieve vitrification, and then thinned down using a focused ion beam instrument. The resulting slices, typically 100-200 nm-thick, are subsequently transferred to a transmission electron microscope (TEM), while maintaining temperatures close to that of liquid nitrogen (77 K). In the TEM, the cells are imaged from different directions by tilting them within the microscope. The resulting series of images is computationally reconstructed to yield a 3D tomogram capturing a snapshot of the cell interior at molecular resolution.
Guo et al. have used cryo-ET to zoom into neuronal poly-GA aggregates similar to those found in the brain of C9ORF72 ALS/FTD patients. They discovered that these aggregates consist of a dense network of twisted ribbons, together with an unexpected guest. Guo et al. realized that in between the poly-GA ribbons there was a dramatic accumulation (over 30 times normal levels) of a critical macromolecular complex known as the “proteasome” (Figure 1).
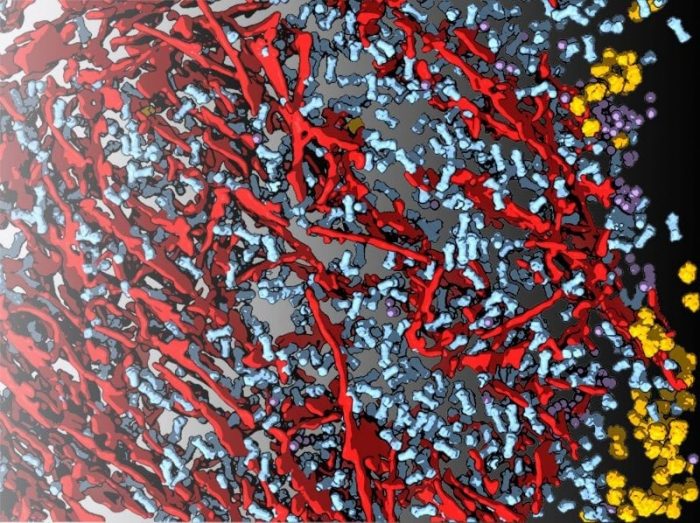
Figure 1: Neuronal C9ORF72 poly-GA aggregates imaged in situ by cryo-electron tomography. In neurons, C9ORF72 poly-GA aggregates are formed by ribbons (red) that sequester a large number of 26S proteasomes (blue). Other macromolecules such as ribosomes (yellow) and the TRiC chaperonin (purple) are found mainly in the periphery of the aggregates. Credit: Qiang Guo / Max Planck Institute of Biochemistry.
In normal cells proteasomes are in charge of waste disposal and recycling, as they degrade dysfunctional proteins and reduce them to their basic components for further re-use in the cell. However, in neurons expressing poly-GA, a significant fraction of the proteasomes was sequestered at the aggregates. Biochemical and structural analysis at unprecedented resolution showed that poly-GA-associated proteasomes were functionally impaired, indicating that poly-GA aggregation significantly alters the cellular systems of protein homeostasis. Therefore, the formation of poly-GA aggregates clogs a critical waste disposal system of the neuron, compromising the health of these cells.
These results are in line with previous literature describing how the GA-rich sequence of the Epstein-Barr virus (EBV) nuclear antigen 1 protein also hinders proteasomal degradation. This polypeptide seems to enter the proteasome, but fails to be degraded and gets stuck (Kraut, 2013). Interestingly, it has been proposed that EBV, which causes infectious mononucleosis and is associated with lymphoid and epithelial tumors, employs this mechanism to escape cellular surveillance and establish latent infections (Masucci, 2004). Therefore, the study of Guo et al. establishes a surprising link between the pathology of EBV infection and neurodegeneration in C9ORF72 ALS/FTD.
Although protein aggregation and proteasome dysfunction are common features of many neurodegenerative conditions (Dantuma and Bott, 2014; Hipp et al., 2014), poly-GA aggregates were substantially different from those related to e.g. Huntington’s disease, in which proteasomes did not accumulate (Bauerlein et al., 2017). This suggests that the contribution of aggregates to neurodegeneration is different in each disease (Pontano Vaites and Harper, 2018) and that further research is needed to unravel this tantalizing diversity.
These findings are described in the article entitled In Situ Structure of Neuronal C9orf72 Poly-GA Aggregates Reveals Proteasome Recruitment, recently published in the journal Cell. This work was conducted by Qiang Guo, Antonio Martínez-Sánchez, Florian Beck, Manuela Pérez-Berlanga, Frédéric Frottin, Mark S. Hipp, F. Ulrich Hartl, Wolfgang Baumeister, and Rubén Fernández-Busnadiego from the Max Planck Institute of Biochemistry, Carina Lehmer, Hannelore Hartmann, and Dieter Edbauer from the German Center for Neurodegenerative Diseases, and Till Rudack from the Ruhr University Bochum.
References:
- Bauerlein, F.J.B., Saha, I., Mishra, A., Kalemanov, M., Martinez-Sanchez, A., Klein, R., Dudanova, I., Hipp, M.S., Hartl, F.U., Baumeister, W., et al. (2017). In Situ Architecture and Cellular Interactions of PolyQ Inclusions. Cell 171, 179-187 e110.
- Dantuma, N.P., and Bott, L.C. (2014). The ubiquitin-proteasome system in neurodegenerative diseases: precipitating factor, yet part of the solution. Front Mol Neurosci 7, 70.
- DeJesus-Hernandez, M., Mackenzie, I.R., Boeve, B.F., Boxer, A.L., Baker, M., Rutherford, N.J., Nicholson, A.M., Finch, N.A., Flynn, H., Adamson, J., et al. (2011). Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245-256.
- Edbauer, D., and Haass, C. (2016). An amyloid-like cascade hypothesis for C9orf72 ALS/FTD. Curr Opin Neurobiol 36, 99-106.
- Guo, Q., Lehmer, C., Martinez-Sanchez, A., Rudack, T., Beck, F., Hartmann, H., Perez-Berlanga, M., Frottin, F., Hipp, M.S., Hartl, F.U., et al. (2018). In Situ Structure of Neuronal C9orf72 Poly-GA Aggregates Reveals Proteasome Recruitment. Cell 172, 696-705 e612.
- Hipp, M.S., Park, S.H., and Hartl, F.U. (2014). Proteostasis impairment in protein-misfolding and -aggregation diseases. Trends Cell Biol 24, 506-514.
- Kraut, D.A. (2013). Slippery substrates impair ATP-dependent protease function by slowing unfolding. J Biol Chem 288, 34729-34735.
- Masucci, M.G. (2004). Epstein-Barr virus oncogenesis and the ubiquitin-proteasome system. Oncogene 23, 2107-2115.
- Pontano Vaites, L., and Harper, J.W. (2018). Protein aggregates caught stalling. Nature 555, 449-451.
- Renton, A.E., Majounie, E., Waite, A., Simon-Sanchez, J., Rollinson, S., Gibbs, J.R., Schymick, J.C., Laaksovirta, H., van Swieten, J.C., Myllykangas, L., et al. (2011). A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72, 257-268.
- Wagner, J., Schaffer, M., and Fernandez-Busnadiego, R. (2017). Cryo-electron tomography-the cell biology that came in from the cold. FEBS Lett 591, 2520-2533.
Related Posts
 Important Strategies To Help Healthcare Providers Support Patients With Diabetes
Important Strategies To Help Healthcare Providers Support Patients With Diabetes Studying The Link Between Increased BMI And Late-Onset Preeclampsia In Pregnant Women
Studying The Link Between Increased BMI And Late-Onset Preeclampsia In Pregnant Women A Bacterial Cell Imaging Method Using CRISPR And Microfluidics
A Bacterial Cell Imaging Method Using CRISPR And Microfluidics B. infantis Reduces Key Markers Of Intestinal Inflammation In Infants
B. infantis Reduces Key Markers Of Intestinal Inflammation In Infants Epigenetic Changes In Multiple Sclerosis – Studied In Twins
Epigenetic Changes In Multiple Sclerosis – Studied In Twins Did You Know Math Can Help Us Learn How Diseases Work?
Did You Know Math Can Help Us Learn How Diseases Work?