





Our immune system protects us from diseases and infections. It is made up of a complex network of cells and organelles, communicating and functioning together to recognize and tackle any invading foreign substance (antigen) that may or may not threaten our wellbeing. The immune system has a unique ability to recognize and remember multitudes of different antigens.
Additionally, it has the capability to distinguish between body’s own (“self”) and foreign (“non-self”) antigens, enabling it to act specifically against only foreign invaders, without harming one’s own body components. This feature is an important aspect of immune regulation achieved through continuous programming of B-cells and T-cell lymphocytes), by repetitive cycles of selection and elimination whereby self-tolerant immune cells are selected and self-reactive immune cells are eradicated during the initial stage of development and differentiation (Figure 1).
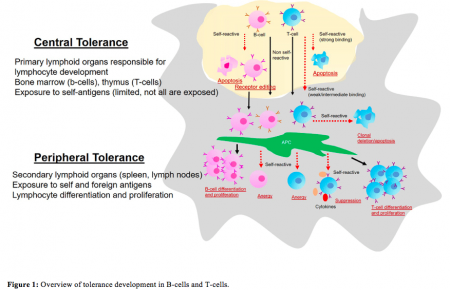
Credit: B. Vijayalakshmi Ayyar
Malfunctioning in this programming stage causes the immune cells to fail to differentiate between self and foreign leading it to attack one’s own system resulting in autoimmunity. The exact mechanism that triggers autoimmunity is unknown, however, genetic factors are known to play a key role in pre-disposing one to acquire autoimmune disorders. There are more than 80 types of autoimmune diseases among which our lab focused on myasthenia gravis (MG).
MG is a T-cell-mediated autoimmune disorder caused by the presence of autoantibodies to acetylcholine receptor (AchR) – or muscle-specific tyrosine kinase (MuSK) receptors, responsible for neuromuscular transmission. MG is characterized by muscle weakness and rapid fatigue caused by a breakdown in the normal communication between nerves and muscles. Majority of the MG treatments are non-specific, owing to the poorly understood pathogenesis of the disease; complicating the search for more specific and efficacious treatment options.
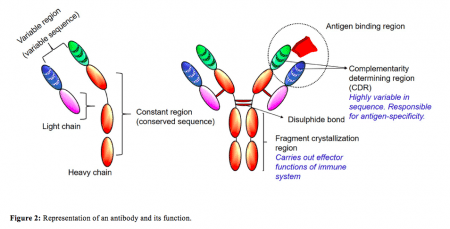
Credit: B. Vijayalakshmi Ayyar
Extensive research showed that certain genetic factors are associated with MG. Based on this data, we devised our strategy to selectively inhibit AChR-reactive T-cell activation by blocking disease-related DQB1 alleles (genetic factor). For this purpose, we developed a set of mouse monoclonal antibodies (mAbs) specific to MG-associated disease-related DQB1 alleles. These antibodies were capable of inhibiting disease-specific T-cell proliferation when used in an in vitro assay using T-cells from MG-patients, showing their promise as a therapeutic.
As the antibodies were developed in mouse, they were not suitable for human treatment due to their foreign nature. To overcome this, the current study was designed to humanize an MG-specific monoclonal antibody by switching the mouse sequence with human sequence, thus, making it more acceptable for treatment. However, antibodies have complex structures with different segments accountable for different functions (Figure 2).
We were only interested in the antigen-binding region of the antibody so we engineered our antibody to a single chain fragment variable (scFv) by grafting the antigen binding parts of the mouse antibody to a suitable human antibody framework (Figure 3). Antibody engineering and humanization is a complicated process which needs to be performed in a systematic manner to avoid issues with the expression and functional properties of the antibodies. The current study details the stepwise method in which a full-length antibody was engineered and humanized. The humanized scFv was then tested, in vitro, for efficacy comparing it with the parent mouse antibody. This is the first report of a humanized scFv development as a possible therapeutic for MG.
These findings are described in the article entitled Development of humanized scFv antibody fragment(s) that targets and blocks specific HLA alleles linked to myasthenia gravis, published in the journal Applied Microbiology and Biotechnology. This work was led by B. Vijayalakshmi Ayyar from Baylor College of Medicine.
Related Posts
 Important Strategies To Help Healthcare Providers Support Patients With Diabetes
Important Strategies To Help Healthcare Providers Support Patients With Diabetes Studying The Link Between Increased BMI And Late-Onset Preeclampsia In Pregnant Women
Studying The Link Between Increased BMI And Late-Onset Preeclampsia In Pregnant Women A Bacterial Cell Imaging Method Using CRISPR And Microfluidics
A Bacterial Cell Imaging Method Using CRISPR And Microfluidics B. infantis Reduces Key Markers Of Intestinal Inflammation In Infants
B. infantis Reduces Key Markers Of Intestinal Inflammation In Infants Epigenetic Changes In Multiple Sclerosis – Studied In Twins
Epigenetic Changes In Multiple Sclerosis – Studied In Twins Did You Know Math Can Help Us Learn How Diseases Work?
Did You Know Math Can Help Us Learn How Diseases Work?