




Our genome has two copies of each gene, contributed to equally by our mother and father. Having two copies of each gene allows for compensation if one of the copies has a mutation that renders it non-functional. However, some of the genes in our genome absolutely need two working copies for normal function and having only one non-functional copy can cause disease, technically referred to as “haploinsufficiency.”
It is currently estimated that over 660 genes lead to disease due to haploinsufficiency. Gene therapy could be a potential treatment for haploinsufficiency where extra copies of a functional gene can be delivered into the body to compensate for the loss of gene function. Recombinant adeno-associated virus (AAV) is a promising therapeutic choice as a delivery vehicle for gene therapy, as it provides long-lasting gene expression, delivers DNA without integrating into the genome, and has limited pathogenicity. Due to that, several AAV-based gene therapies to treat genetic disorders are being developed, and some are already being used in the clinic. However, the packaging capacity of adeno-associated virus limits its use to deliver transgenes that have longer coding DNA sequences.
To circumvent the limitation of AAV packaging capacity and provide a precise gene therapy approach, we took advantage of the existing normal copy. The endogenous expression of a gene is regulated by specific DNA elements called cis-regulatory elements. The promoter is a cis-regulatory element that resides right next to the gene, and enhancers reside further away and regulate when, where, and at what levels the promoter is turned on.
We hypothesized that if we can target these cis-regulatory elements in such a way that extra copies of mRNA are made from the existing functional copy, we could compensate for the haploinsufficiency of the gene. To do this, we chose a CRISPR-based system that has a mutant Cas9 that cannot cut the DNA (dead dCas9) but can target a precise genomic location and can bring along for the ride a transcriptional activator to increase expression of a target gene. We used a fusion of nuclease deficient dead-Cas9 with a generic transcriptional activator domain derived from the viral protein 16 (VP64) in combination with single guide RNA (sgRNA) to target and activate the promoter or enhancer of a haploinsufficient gene/s. We decided to use a haploinsufficient mouse model for monogenic obesity as a proof of principle of this approach, as it is an easy quantifiable trait to measure.
We tested CRISPR mediated activation (CRISPRa) of two genes single-minded homolog 1 (SIM1) and melanocortin 4 receptor (MC4R). Both SIM1 and MC4R are expressed in the hypothalamus and regulate food-intake and body metabolism. Mutations in both these genes that lead to haploinsufficiency are a common cause of monogenic obesity in humans, leading to excessive eating. Haploinsufficient mouse models of Sim1 and Mc4r also show hyperphagia and excess body weight gain.
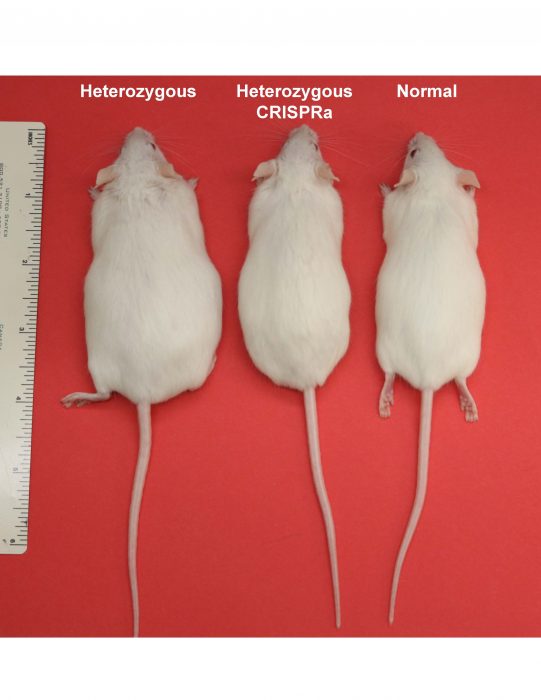
Image courtesy Nadav Ahituv
We generated a CRISPRa system in transgenic mice and tested if the obesity phenotype of Sim1 haploinsufficient mice can be rescued by it. We designed CRISPRa that can target the promoter or enhancer of Sim1 to upregulate Sim1 expression from the functional copy. We measured body weight of these mice up to 16 weeks. We observed a complete rescue of the obesity phenotype in Sim1 haploinsufficient mice both by targeting CRISPRa to the promoter or enhancer of Sim1. Sim1 haploinsufficient mice that carry a transgene of CRISPRa did not exhibit hyperphagia and accumulated less body fat. We found rescue of obesity in both groups of mice that carry either Sim1 promoter CRISPRa or Sim1 enhancer CRISPRa. Interestingly, we found that Sim1 upregulation was achieved only in tissues where the targeted cis-regulatory element is active despite the ubiquitous expression of CRISPRa system. Targeting the Sim1 promoter with CRISPRa upregulated Sim1 in all the tissues where it is normally expressed, i.e., kidney and hypothalamus.
When we target the enhancer, which is active only in the hypothalamus, CRISPRa upregulated Sim1 expression only in the hypothalamus. Previous studies have shown that the CRISPR system can have off-target binding. We assessed its off-target effects in these animals in two ways; 1) We looked for any abnormal gene expression in the hypothalamus via RNA-seq; 2) We checked for any off-target CRISPRa binding in the genome via ChIP-seq. We did not find any off-targeting CRISPRa binding in the genome that can result in atypical gene expression.
These results were quite encouraging and motivated us to try CRISPRa correction in post-natal mice. We designed and packaged our CRISPRa system in AAV (CRISPRa-AAV) and injected it into the hypothalamus of haploinsufficient Sim1 mice at 4 weeks of age. We observed these mice for up to 10 months and noticed that they did not gain excessive body weight, compared to Sim1 haploinsufficient controls. To show that our approach can work on additional genes, we next used haploinsufficient Mc4r mice that have one functional copy of Mc4r and are genetically pre-determined to gain excessive body weight. We replicated our CRISPRa strategy targeting the promoter of Mc4r and showed that we can rescue their obesity phenotype compared to haploinsufficient Mc4r control mice.
This study provides a proof-of-concept in an animal model system that CRISPRa can be developed as a potential therapeutic tool to treat haploinsufficiency. As there are 660 genes that are known to lead to human disease due to haploinsufficiency, CRISPRa could be used as a potential approach to treat them. Future work will be required to address the safety and toxicity of this approach. Since this approach is both CRISPR and AAV based, thorough analysis of pathogenicity and immunogenicity is needed.
We should note that there are some substantial advantages and merits to this strategy: 1) It does not create any mutation or edits to the DNA; 2) it takes advantage of the gene regulatory mechanism of the gene itself thus limiting the ectopic gene expression that can be observed in conventional gene therapy; 3) Most importantly, CRISPRa can be used to upregulate a haploinsufficient gene that cannot be packaged into AAV vectors due to packaging size limitations. This size limitation is currently prohibitive for around 150 out of 660 haploinsufficient disease-causing genes due to their longer coding DNA sequence, but CRISPRa packaged into AAV can potentially be used to rescue these diseases.
In summary, this study highlights the potential of using CRISPRa as a therapeutic tool for diseases caused by gene dosage and future studies are required to explore the full therapeutic potential of this approach.
These findings are described in the article entitled CRISPR-mediated activation of a promoter or enhancer rescues obesity caused by haploinsufficiency, recently published in the journal Science.
Related Posts
 Important Strategies To Help Healthcare Providers Support Patients With Diabetes
Important Strategies To Help Healthcare Providers Support Patients With Diabetes Studying The Link Between Increased BMI And Late-Onset Preeclampsia In Pregnant Women
Studying The Link Between Increased BMI And Late-Onset Preeclampsia In Pregnant Women A Bacterial Cell Imaging Method Using CRISPR And Microfluidics
A Bacterial Cell Imaging Method Using CRISPR And Microfluidics B. infantis Reduces Key Markers Of Intestinal Inflammation In Infants
B. infantis Reduces Key Markers Of Intestinal Inflammation In Infants Epigenetic Changes In Multiple Sclerosis – Studied In Twins
Epigenetic Changes In Multiple Sclerosis – Studied In Twins Did You Know Math Can Help Us Learn How Diseases Work?
Did You Know Math Can Help Us Learn How Diseases Work?