





Deposition of amyloid beta (Aβ) plaques in the brain is the most acknowledged feature of Alzheimer’s disease. Despite decades of research, debate persists over the impact of plaques on neuronal activity and brain function.
At the core of the amyloid plaque controversy, which also overshadows the amyloid hypothesis of Alzheimer’s disease, lie three sets of observations: (1) a large number of cases of overt dementia in elderly without Aβ plaques; (2) numerous accounts of excessive load of Aβ plaques in brain without dementia or cognitive deficit; and (3) emerging evidence for important physiological role of Aβ in regulating neural homeostasis and synaptic functions.
Indeed, Aβ seems to regulate multiple processes at synaptic terminals, from activation of voltage-activated calcium influx to trafficking and fusion of synaptic vesicles and post-exocytose recovery. Intriguingly, under certain circumstances, Aβ plaques have been also suggested to play a neuroprotective role by sequestering toxic Aβ species from their surrounding environment.
The widening gap between the extent of amyloid load in the brain and clinical manifestation of Alzheimer’s disease, thus, leave neuroscientists to wonder if and how amyloid plaques are related to one of the most complex diseases affecting the brain and mind. In this regard, a very useful piece of information has come from studies showing that amyloid plaques are not specific to Alzheimer’s disease, but occur also in other neurological diseases, including spongiform encephalopathies, traumatic brain injuries, neuro-syphilis, and, occasionally, also in the normal aging brain. Hence, the deposits of Aβ in the brain per se do not warrant neurological and psychiatric signs of Alzheimer’s disease.
Another critical piece of information emerged recently from reports showing a highly complex and localized response of brain tissue to amyloid plaques. It seems that the development of plaques is far from an unpretentious and gradual peptide deposition in brain tissue but involves highly complex local reactions contributed by an array of cellular and molecular players. Indeed, there seem to be highly diverse plaque-related changes, with a considerable amount of axons and neurites undergoing sprouting, swellings, and dystrophic alterations. But what do all these changes have to do with clinical manifestations of Alzheimer’s disease?
First and foremost, glutamatergic neurons, which are the key drivers of brain activity, seem to be the most responsive neuron type to amyloid lesions and display massive swellings and dystrophic changes. Secondly, the swollen and dystrophic glutamatergic axons around amyloid lesions become enriched with canonical pre-synaptic proteins and small translucent vesicles, which in the normal brain are strictly co-localized at the axon terminals, specialized for transmitter release. Thus, it appears that to some glutamatergic neurons, amyloid lesions present effective cues activating synaptogenesis, which initiates complex molecular changes that lead to the development of molecular and cellular scaffolds capable of releasing glutamate.
Critically, all these rearrangements in axons occur in the absence of postsynaptic elements or specialized mechanisms for containing the released extracellular glutamate. Collectively, these alterations render axonal dystrophies around amyloid plaques sites for uncontrolled leakage of glutamate, which leads to a raising of its level with potent stimulator effects on neuronal activity.
While not yet fully appreciated, the ectopic release of glutamate seems to have powerful local and extended effects on neural activity. And this is hardly surprising given that the maintenance of exquisite neurochemical balance, and especially glutamate levels, is critical for normal brain functions.
Accordingly, the damage caused by amyloid plaques may be not due to Aβ peptide per se but arrive as a consequence of non-physiological response of neurons to plaques, with a strong increase in the activity of non-synaptic glutamate in pockets surrounding plaques. The highly variable reaction of neurons and brain tissue to amyloid plaques on one side, thus, seems to explain at least in part, numerous cases of dissociation between the amyloid pathology and cognitive deficit with memory loss. On the other, free-floating glutamate in the brain is highly toxic and can cause extensive disruptions of neuronal activity with progressive loss of synaptic connections, and initiate the neurodegeneration.
With growing evidence for early onset functional abnormalities in cortical circuits of Alzheimer’s disease, the abnormal glutamatergic drive warrants careful future research. In addition to settling the controversy over the role of plaques in the pathobiology of Alzheimer’s disease, the outcome of upcoming studies could afford major advances towards developing a new perspective on brain functions and disease, an investment holding major research and translational benefits.
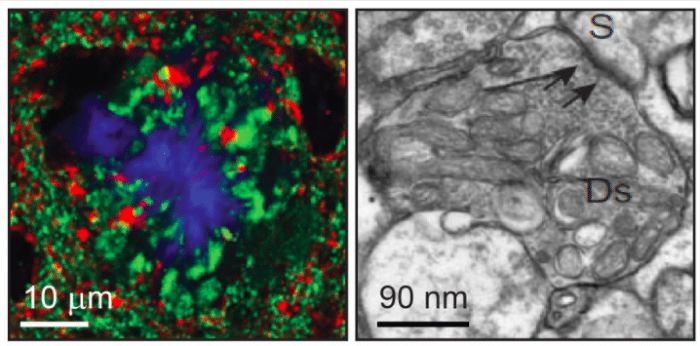
FIG. 1: Left: confocal micrograph of amyloid plaque in Alzheimer’s disease mouse model brain (blue) with axonal dystrophies enriched with synaptic vesicles (green). Right: electron micrograph of dystrophic axons containing small synaptic vesicle (black arrows). Courtesy by Ovsepian et al. 2017 https://academic.oup.com/cercor/article/27/10/4733/3056434, republished with permission from the publisher.
These findings are described in the article entitled Amyloid Plaques of Alzheimer’s Disease as Hotspots of Glutamatergic Activity, recently published in the journal Neuroscientist. This work was conducted by Saak V. Ovsepian and Vasilis Ntziachristos from the German Research Center for Environmental Health and the Technical University of Munich, Laszlo Zaborsky from Rutgers University, and Valerie B. O’Leary and J. Oliver Dolly from Dublin City University.
References:
- Ovsepian et al., Ambient Glutamate Promotes Paroxysmal Hyperactivity in Cortical Pyramidal Neurons at Amyloid Plaques via Presynaptic mGluR1 Receptors. Cereb Cortex. 2017 Oct 1;27(10):4733-4749. doi: 10.1093/cercor/bhw267.
- Ovsepian et al., Amyloid Plaques of Alzheimer’s Disease as Hotspots of Glutamatergic Activity. Neuroscientist. 2018 Jul 27:1073858418791128. doi: 10.1177/1073858418791128.
Related Posts
 Genes Keep You On A Long Leash With Your Dog
Genes Keep You On A Long Leash With Your Dog Looking At Versus Focusing On Faces: What Attracts Our Attention?
Looking At Versus Focusing On Faces: What Attracts Our Attention? What Do They Get Out Of It? The Emotional Experience Of Providing Social Support
What Do They Get Out Of It? The Emotional Experience Of Providing Social Support How Does Rewarding Safe Choices Affect Teen Decision Making In Peer Contexts?
How Does Rewarding Safe Choices Affect Teen Decision Making In Peer Contexts? Adolescent Self-Efficacy Is Shaped By Family, School, And Peers
Adolescent Self-Efficacy Is Shaped By Family, School, And Peers Does Morality Make Us Make God In Our Own Image?
Does Morality Make Us Make God In Our Own Image?